
CE / CME
Emerging Data and Clinical Implications in Multiple Myeloma: ASH 2025 Highlights
Physician Assistants/Physician Associates: 1.00 AAPA Category 1 CME credit
Pharmacists: 1.00 contact hour (0.1 CEUs)
Physicians: maximum of 1.00 AMA PRA Category 1 Credit™
Nurse Practitioners/Nurses: 1.00 Nursing contact hour
Released: March 05, 2026
Expiration: September 04, 2026


Activity
MajesTEC-3: Teclistamab + Daratumumab vs Daratumumab-Based Regimens (DPd/DVd) in Patients With R/R MM
Sagar Lonial, MD
Let’s start off talking about MajesTEC-3, which was one of the really important abstracts presented at ASH 2025. It was in the plenary session, and it was also published in the New England Journal of Medicine at the same time as its initial presentation.1,2 MajesTEC-3 was a randomized, phase III trial of teclistamab plus daratumumab vs daratumumab-based standard of care (SoC) regimens, which ended up being almost predominantly daratumumab, pomalidomide, and dexamethasone.
Sagar Lonial, MD
The rationale behind this approach relates to the fact that 1 of the challenges with bispecific antibodies is that their activity may be limited by the presence of CD38+ immunosuppressive or T-regulatory cells. However, daratumumab not only provides single-agent activity but also eliminates immunosuppressive CD38+ cells in the tumor microenvironment.3,4 This could perhaps make the BCMA bispecific—in this case, teclistamab—more effective.
Sagar Lonial, MD
The randomized, phase III MajesTEC-3 trial enrolled patients with R/R MM with 1-3 prior lines of therapy. A total of 587 patients were randomly assigned to teclistamab plus daratumumab, with teclistamab basically following the daratumumab schedule, or an SoC regimen, either daratumumab, DPd, or DVd. These schedules were fairly standard as well.
Those are 2 very reasonable regimens for the control that are used quite commonly. The primary endpoint was PFS per independent review committee; secondary endpoints included response rates, MRD, and OS.1,2
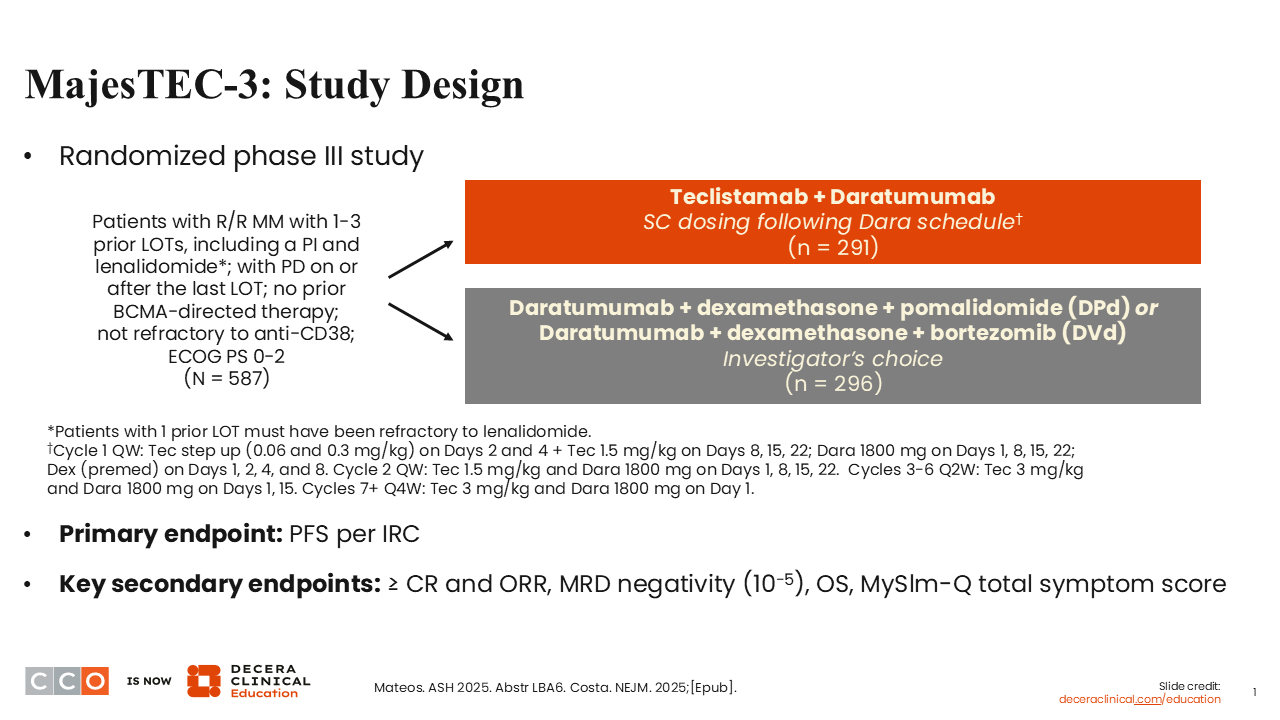
MajesTEC-3: Baseline Characteristics
Sagar Lonial, MD
Looking at patient characteristics, the benefit of a nice randomized, phase III trial is that you get balance between the arms.1,2 The numbers of pretty much every metric are similar between arms. A small number of patients had extramedullary plasmacytomas—approximately 5% in each arm. We will discuss the management of those patients in the next study. Certainly, in this trial, they did not account for anywhere near the majority of patients evaluated.
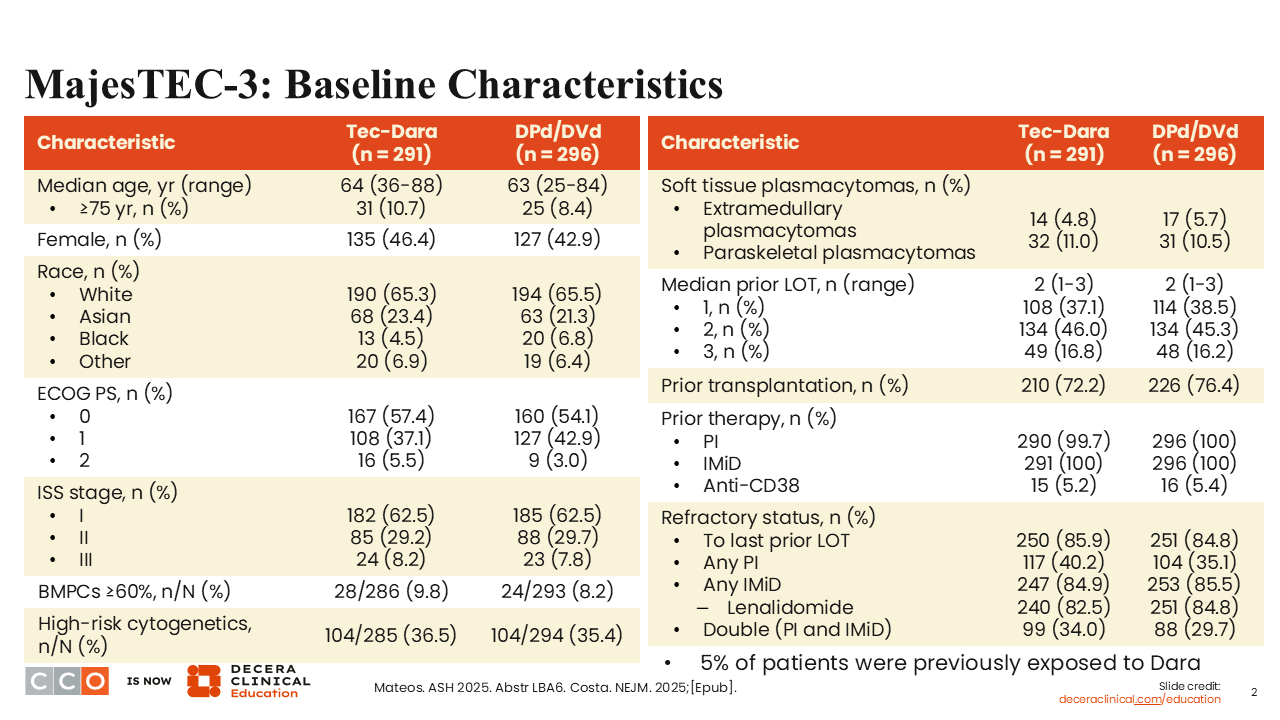
MajesTEC-3: PFS
Sagar Lonial, MD
I think 2 things were really quite striking. First, the 36-month PFS of 83.4% in the teclistamab plus daratumumab arm is probably the longest PFS we have seen in an early relapse trial.1,2 Just for comparison, because the HRs are always in part related to the efficacy or the effectiveness of the control arm in this setting, DPd performed about as well as we would have expected. I think the control arm did what we would have thought would have been expected from this patient population. The teclistamab plus daratumumab arm did exceptionally well with a long follow-up of 34.5 months. I think these data really do speak to the potency of this pure immune therapy–based combination.
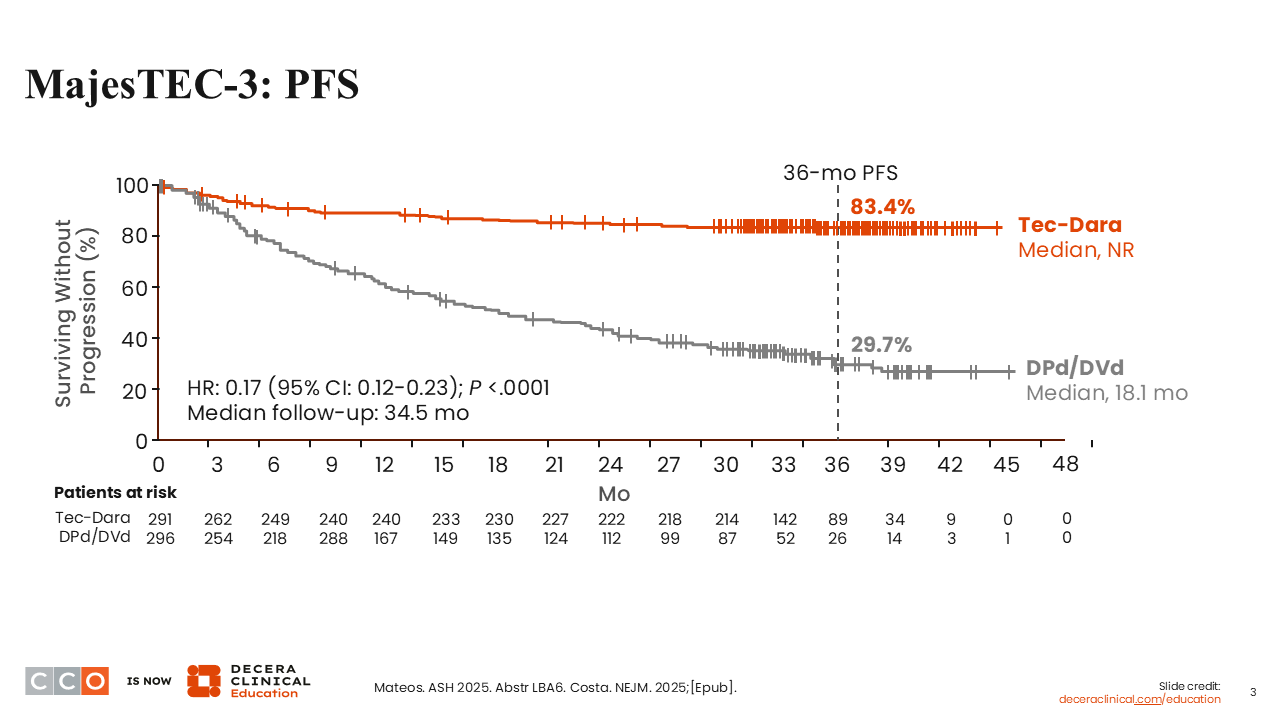
MajesTEC-3: OS
Sagar Lonial, MD
Looking at OS, again, it is rare that you see OS and PFS match as we do in this analysis.1,2 Early on, in the first 6 months, there was a reduction in OS in the teclistamab plus daratumumab arm. That was before the mandatory implementation of monthly IVIG, and there were infections that led to those deaths early on. The use of monthly IVIG appeared to have mitigated that risk over time. I think that is an important take-home message in terms of managing these patients who are getting teclistamab plus daratumumab.
MajesTEC-3: PFS and OS Subgroup Analysis
Sagar Lonial, MD
In the subgroup analysis, pretty much all groups gained significant benefit with teclistamab plus daratumumab as assessed by PFS and OS.1,2 There was some benefit for the group that had baseline plasmacytomas. Certainly, even patients with high-risk genetics gained benefit from the use of teclistamab plus daratumumab almost to the same magnitude as standard-risk patients.
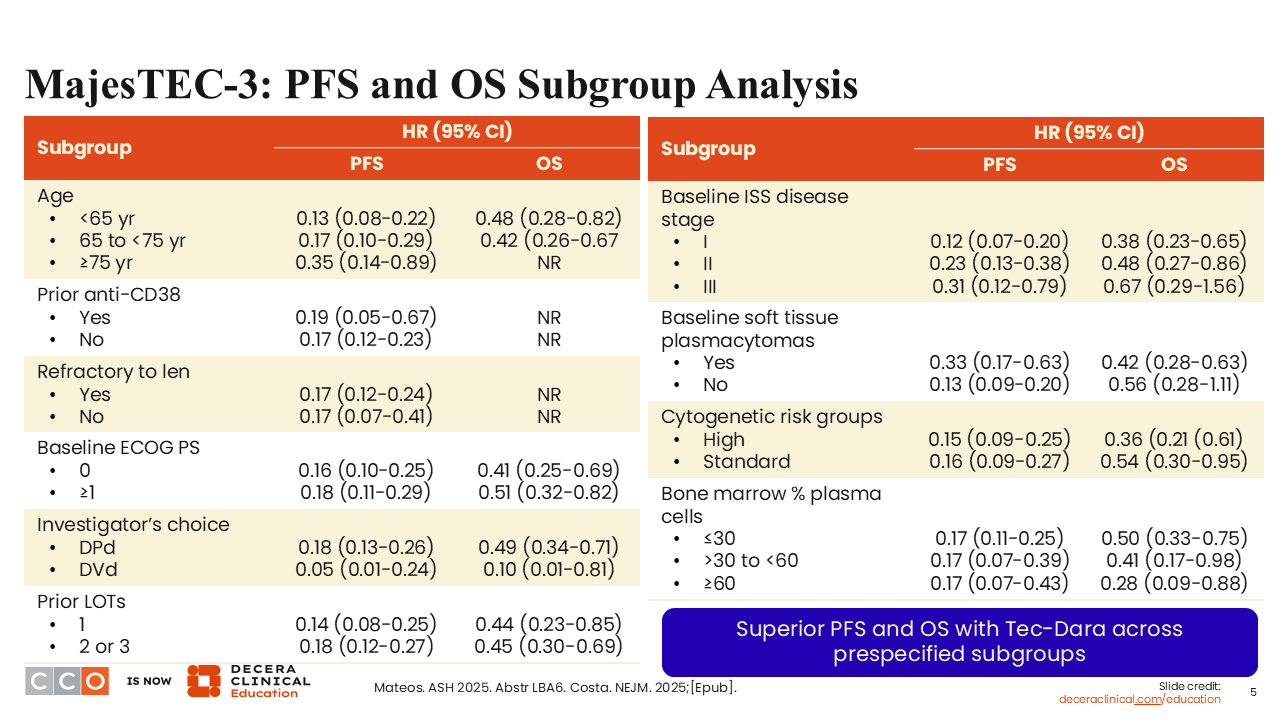
MajesTEC-3: Treatment Response and Duration
Sagar Lonial, MD
In addition to PFS and OS, treatment response and DoR clearly favored the teclistamab plus daratumumab arm.1,2
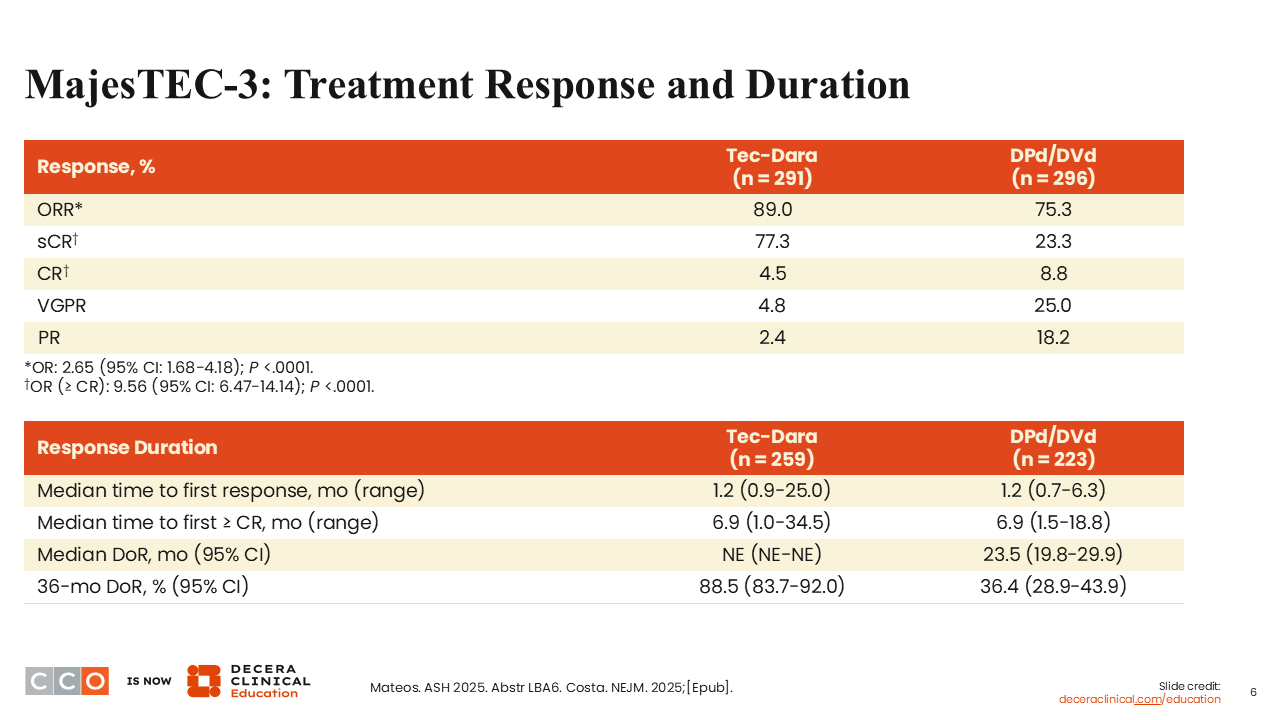
MajesTEC-3: MRD Negativity
Sagar Lonial, MD
I think this represents one of the largest PFS benefits we have ever seen in an early relapse patient population, and that was reflected by MRD negativity outcomes.1,2 You can see big differences between teclistamab plus daratumumab and SoC at both 10-5 and at 10-6, with 53.8% of patients achieving MRD negativity in the teclistamab plus daratumumab arm, compared with only 10.4% in the DPd/DVd arm at the higher sensitivity.
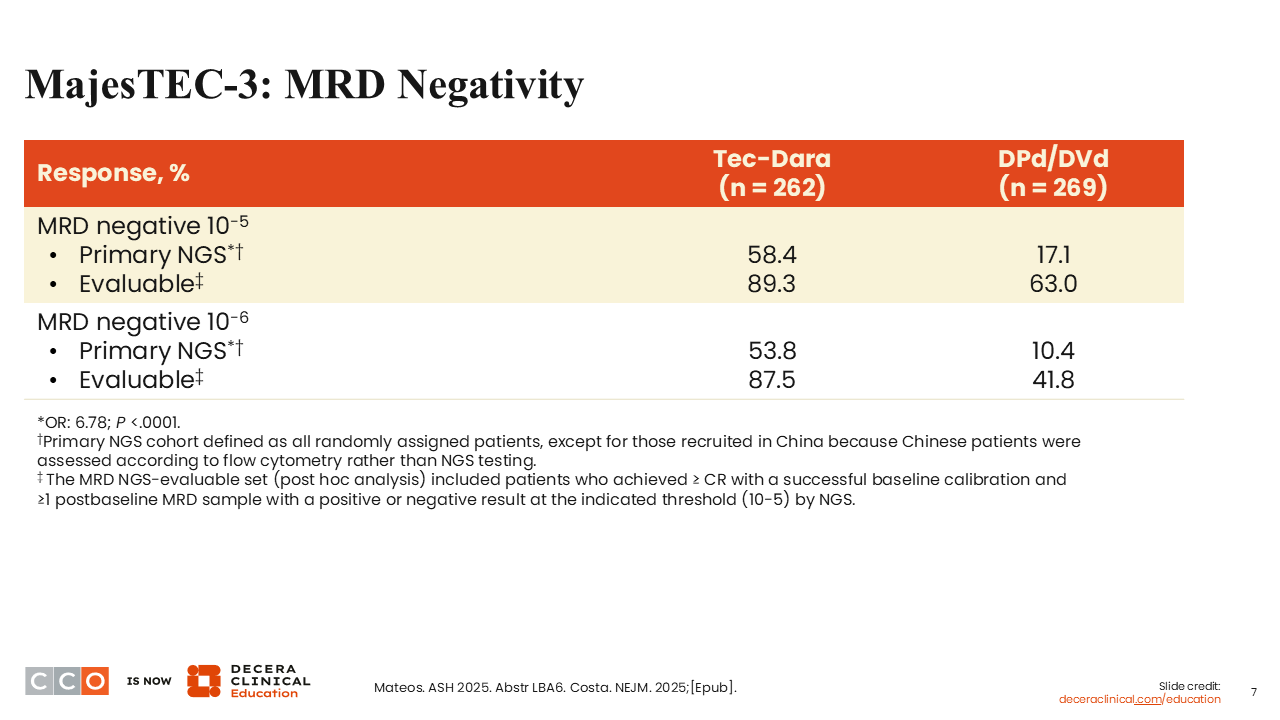
MajesTEC-3: Safety
Sagar Lonial, MD
Regarding safety, infections really represented the majority of what I think we need to be aware of.1,2 You can see there was some neutropenia, of similar magnitude to what we saw with DPd/DVd. Cytokine release syndrome (CRS) occurred in 60.1% of patients, which was not unexpected, and the incidence of diarrhea was higher with teclistamab plus daratumumab vs DPd/DVd, at 51.9% and 30.7%, respectively. Rates of cough and fever were also higher with teclistamab plus daratumumab. The rate of serious AEs was 70.7% with teclistamab plus daratumumab vs 62.4% with DPd/DVd. There was a slightly higher death rate in the teclistamab plus daratumumab arm (7.1% vs 5.9%), again, likely related to that early death before mandatory IVIG administration.
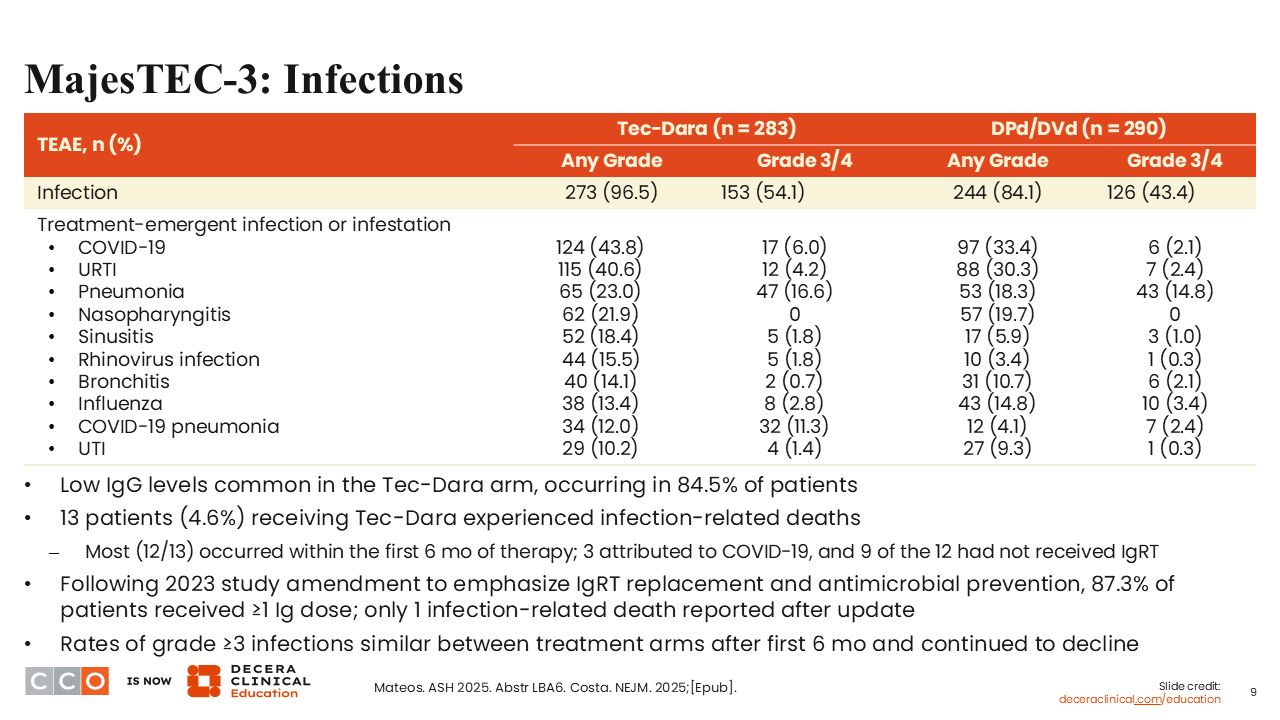
MajesTEC-3: Infections
Sagar Lonial, MD
Here is a breakdown of infections.1,2 Clearly, there is an infection signal with teclistamab plus daratumumab, so vigilance, prophylactic empiric antibiotics, and immunoglobulin replacement therapy are really important to making this a safer and more tolerated regimen.
Conclusions
Shaji Kumar, MD
This is one of the best results we have seen in that first relapse setting. Clearly, the reported PFS in MajesTEC-3 is quite long.1,2 I think it is a harbinger of what will be coming, which is the use of this combination in the first-line setting, which hopefully we will be seeing fairly soon.
The biggest challenge I see with the data is the intense monitoring that needs to happen for these patients compared to many of the other regimens that we have been using, especially from the risk of infection standpoint, which needs a significant amount of education.
Now, the trial clearly shows that starting teclistamab and daratumumab gives these results, but it does not tell us how long these treatments need to continue. However, we know from everything we have seen so far that these treatments probably do not need to be continued until disease progression. Understanding when we can stop the treatment, whether it is related to some composite endpoint that integrates the toxicity and the depth of response, is going to be clearly important.
Even when we talk about stopping therapy, again, do we really need both of them to continue, or can we stop 1 first and then stop the other subsequently? I think the other major challenge with the practical implementation of these data is that the majority of the patients in the first relapse will have seen a lot more daratumumab than the patients in this trial have seen. Understanding what is going to be the threshold of previous daratumumab exposure beyond which we may not see the same degree of benefit with this combination is also going to be important.
As Dr Lonial said, very rarely do we see that the high-risk and standard-risk patients get almost similar benefits out of a treatment. When you look at that forest plot from this study, the high-risk and the standard-risk cytogenetic subgroups had the same PFS HR of approximately 0.16. The stage III subgroup appeared to benefit a bit less than the stage I group of patients. Nevertheless, I think with these new modalities, especially with T-cell redirection, we are starting to see some of those traditional prognostic factors not being that relevant anymore.
Sagar Lonial, MD:
You are absolutely right about the questions regarding applicability to the patients who have been exposed to daratumumab. I think certainly for patients who are not on daratumumab maintenance, who only saw daratumumab as part of their induction, the impact is probably less, and I completely agree with you. The questions about schedule, frequency of administration, and when you can discontinue will be important to try to reduce patients' infection risk even further.
RedirecTT-1 Update: Talquetamab + Teclistamab in R/R MM With EMD
Shaji Kumar, MD:
The next study is an update from RedirecTT-1 evaluating talquetamab and teclistamab in patients with R/R MM with EMD.5,6
One of the challenges in myeloma is the treatment of patients who present with EMD, especially related to disease that is totally separate from bone. We often see bony destruction with plasmacytomas that are in continuity with the bone. Here, however, we are talking about EMD that is completely separate from bone.
When patients present with EMD, whether it is at diagnosis or at the time of relapse, their outcomes are relatively poor.7 We have seen over and over from clinical trials that these patients tend to respond to treatment, particularly with some of the newer therapies like immunotherapy, but the response seems to be short lasting.8 We have seen that with bispecific antibodies used as a single agent and also in the context of CAR T-cells.
When the original RedirecTT-1 trial was conducted evaluating the combination of teclistamab and talquetamab—2 bispecific antibodies targeting 2 different antigens on these tumor cells—there was a subgroup of patients who had EMD who seemed to have better outcomes than one would have expected.9 Based on that, a standalone trial was conducted focused only on patients with true plasmacytoma.5,6 This is probably the first time we are doing a trial that is a large trial that is focused specifically on a patient population. In the past, we have predominantly been dependent on subgroup analyses.
This phase II trial essentially looked at the same doses that were used in the RedirecTT-1 trial of talquetamab and teclistamab given every 2 weeks.5,6,9 These were all patients who were triple-class exposed with EMD that had not been irradiated. An important aspect of this study is that patients with nonsecretory or oligosecretory disease were also included. This again is a group of patients who often do not get into clinical trials. The primary goal was to look at overall response rate (ORR); secondary endpoints included PFS.
Another important aspect of this study was its rigorous use of PET scans to ensure that there were responses seen in these plasmacytomas. The Deauville score and the IMPETUS criteria were incorporated into the radiographic criteria for EMD response in addition to the conventional IMWG criteria.5,6

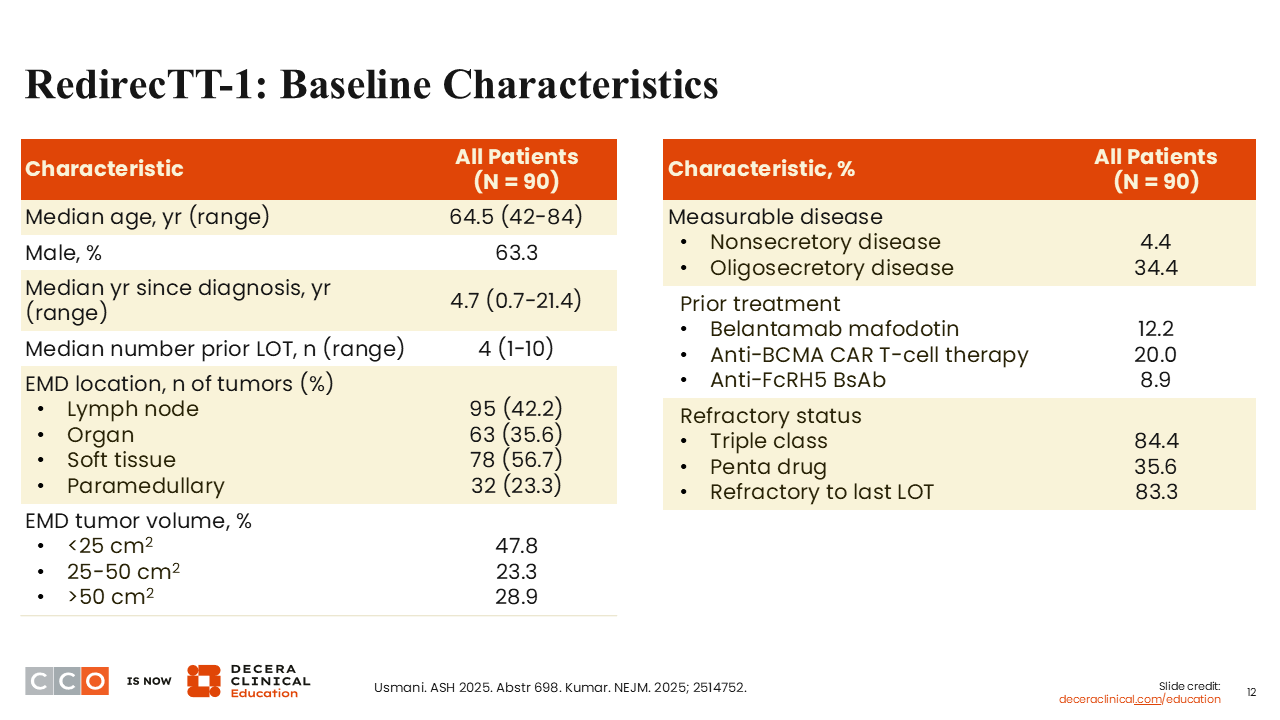
RedirecTT-1: Baseline Characteristics
Shaji Kumar, MD:
The trial enrolled 90 patients. Baseline characteristics were as expected for this patient population. Approximately a third of patients (34.4%) had oligosecretory disease, and 4.4% had nonsecretory disease.5,6 Many of these patients had multiple plasmacytomas in a variety of different soft tissue locations. Patients had received a median of 4 prior lines of therapy. The majority of patients were triple-class refractory (84.4%), and 35.6% were penta-drug refractory. Almost a third of patients had received prior BCMA-targeted therapy, including belantamab mafodotin (12.2%) or CAR T-cells (20.0%).
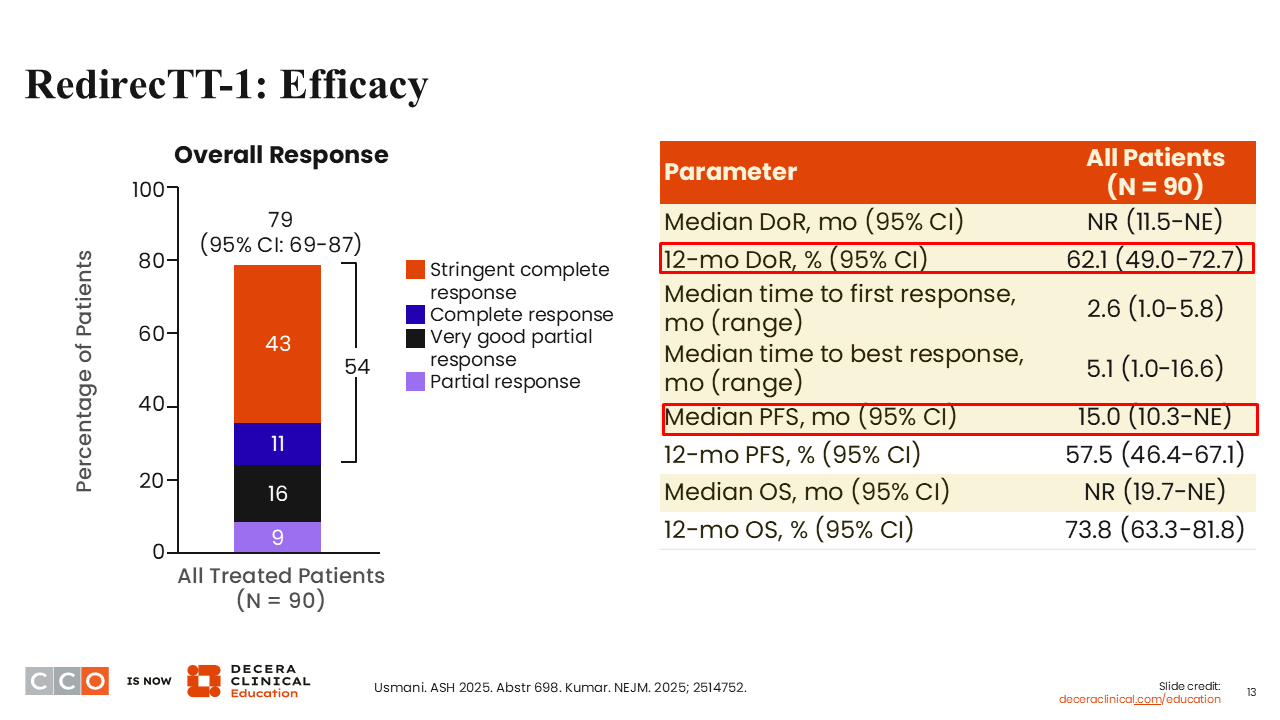
RedirecTT-1: Efficacy
Shaji Kumar, MD:
The ORR with teclistamab and talquetamab was 79%; of more importance, nearly 43% of these patients were in an sCR.5,6 The median PFS was 15.0 months. Nearly two thirds of patients (62.1%) were still in response 12 months after the start of therapy. The median OS was not reached, with almost 75% of the patients (73.8%) alive 1 year from the start of treatment.
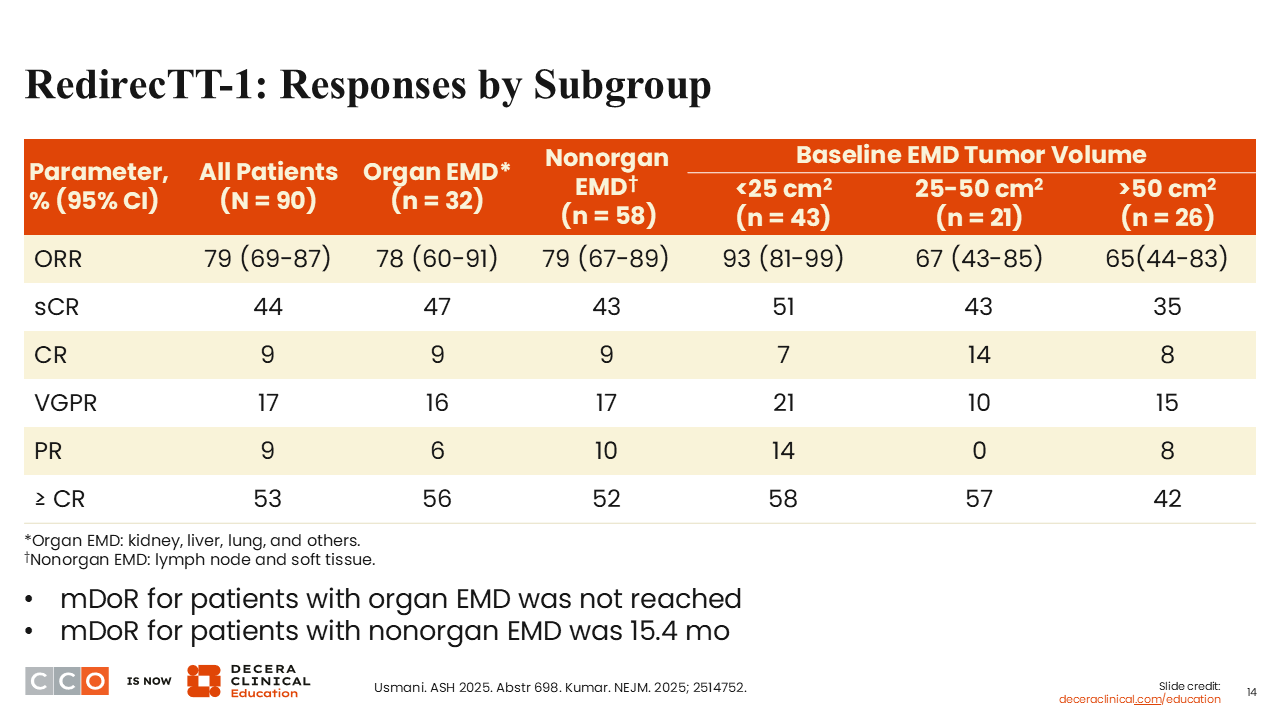
RedirecTT-1: Responses by Subgroup
Shaji Kumar, MD:
Looking at different subgroups, responses were observed in all groups of patients, including in those with organ-based EMD and nonorgan EMD, and irrespective of baseline EMD tumor volume.5,6 Rates of deep responses were also similar across subgroups.
RedirecTT-1: Safety
Shaji Kumar, MD:
The safety profile was quite similar to what we saw with the talquetamab/teclistamab combination in the RedirecTT-1 trial.5,6,9 We see toxicities related to teclistamab and some that are unique to talquetamab, like skin toxicity and mucosal toxicity. Again, rates seemed to be quite similar to those reported in the original trial in terms of the overall proportion of AEs and the proportion of grade ≥3 AEs. There were 11 deaths in the study; 6 were drug-related, and 7 were nonresponders with poor prognosis.
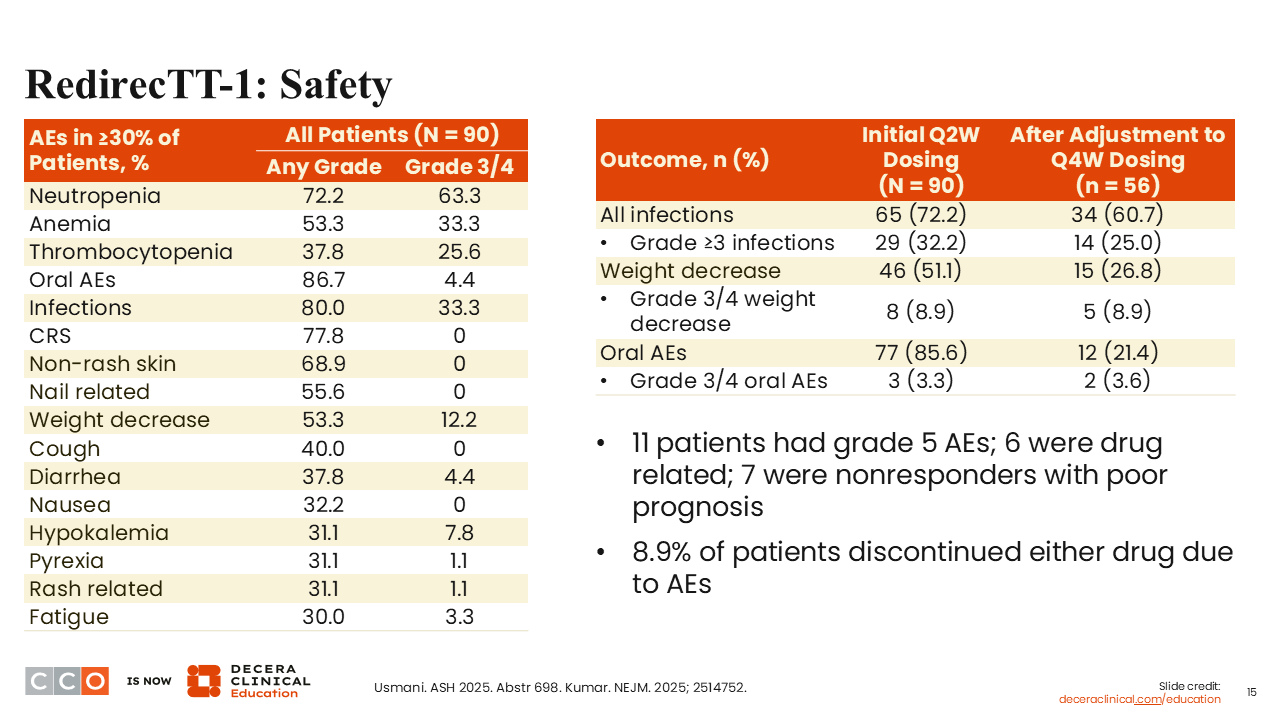
RedirecTT-1: Infections
Shaji Kumar, MD:
One of the concerns with bispecific antibodies is the high rates of infections. Approximately a third of patients in this study developed a grade 3 or 4 infection. Most of these patients had to be on IVIG prophylaxis or replacement therapy. That seemed to have helped, but there were 6 infections that led to death in these patients.
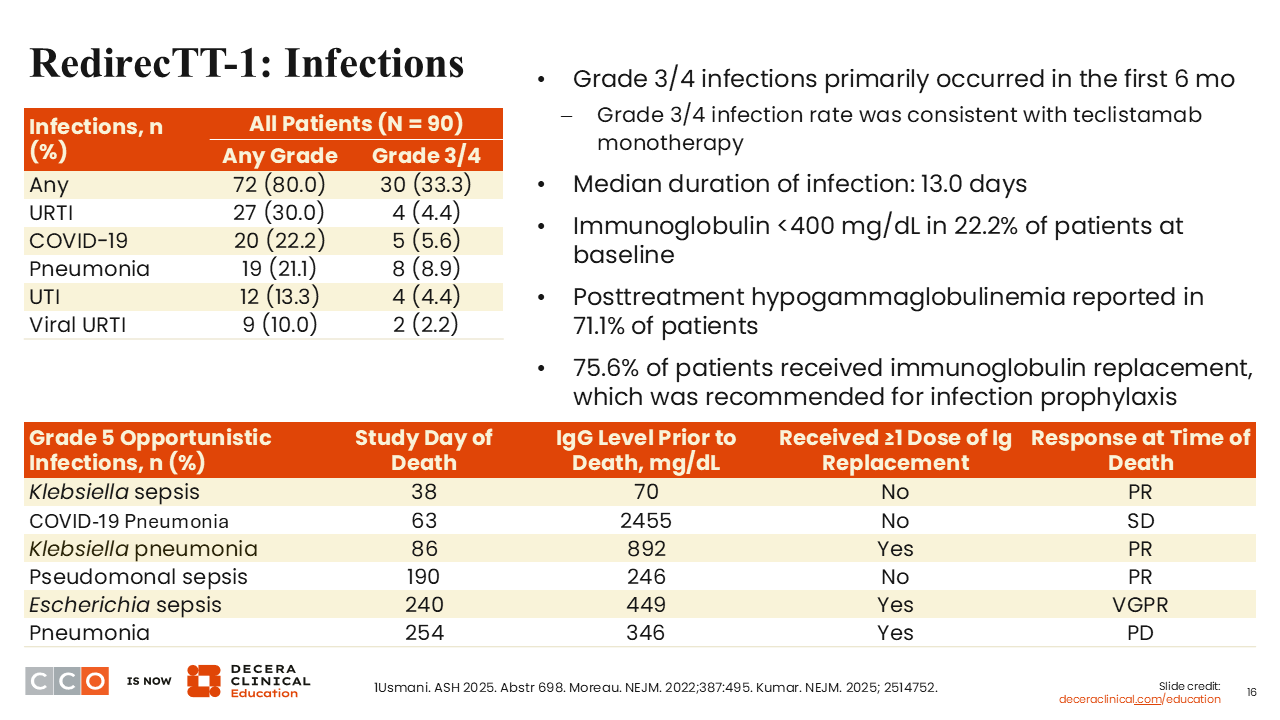
Conclusions
Shaji Kumar, MD:
In these patients with high-risk disease defined by the presence of EMD, the teclistamab/talquetamab combination provided an ORR that was higher than what we have seen previously in this patient population.5,6,8 More important than the response rates is the durability of response, with a median PFS of 15 months. This happens without any additional toxicity compared with what we see with teclistamab and talquetamab individually. Nevertheless, there are toxicities, particularly infections that require very close monitoring and the mucosal and skin toxicities associated with talquetamab.
Sagar Lonial, MD:
The investigators are really to be commended for doing a trial focused almost exclusively on a very challenging group of patients. Seeing these ORR and PFS results in an EMD-based patient population is really quite impressive.
I think the same types of questions that we asked about MajesTEC-3 could also be asked about RedirecTT. How long do you need to give the regimen? How much and how often do you need to give it? What is the optimal prophylaxis or prevention to reduce some of the infectious deaths that we saw in this trial?
Do I think that this combination is suitable for every patient with early relapse disease? Probably not. But for patients with EMD, who are very challenging to treat, I think that this represents a new SoC. I find EMD very difficult to manage. When you get a response, it often does not last very long. The fact that the median PFS is beyond 12 months is, I think, a significant step forward. This really speaks to the value of our new immune therapy approaches in the context of high-risk MM.
CAMMA 1: Updated Safety, Efficacy, and Biomarker Analyses From Phase I Study of Cevostamab + Pomalidomide + Dexamethasone in BCMA1-Naive RRMM
Sagar Lonial, MD:
Next, we are going to talk about the updated safety and efficacy analysis of the phase I CAMMA 1 study evaluating cevostamab plus pomalidomide and dexamethasone in BCMA-naive R/R MM.11
Cevostamab targets FcRH5, yet another immune antigen in myeloma.12 You may hear FcRH5 referred to as FcRL5. Although this is probably immunologically more correct, FcRH5 is more commonly used. Its expression is relatively ubiquitous, similar to BCMA and GPRC5D.13 Unlike GPRC5D, FcRH5 is not expressed on many, if any, nonplasma cells. Consequently, its off-target effects are relatively limited.
We know that FcRH5 targeting with cevostamab is clearly active in R/R MM. That has been presented in a number of different studies by a couple of different groups.13, 14 The concept here was to add in pomalidomide to see whether enhancing immune function could make FcRH5-targeted bispecific antibody therapy even more effective.
Just as a background, we know that this approach works very well. For BCMA-directed therapies, researchers have anecdotally experimented with pomalidomide plus talquetamab and have been able to recapture patients who are progressing on a single-agent bispecific antibody by the addition of pomalidomide, even if the patient's myeloma was resistant to pomalidomide.15 This concept of immune reactivation is a really important one across the board.
This was a relatively straightforward phase 1b study evaluating 2 doses of cevostamab combined with pomalidomide and dexamethasone in BCMA-naive patients who had received at least 1 prior line of therapy and were not refractory to pomalidomide.11

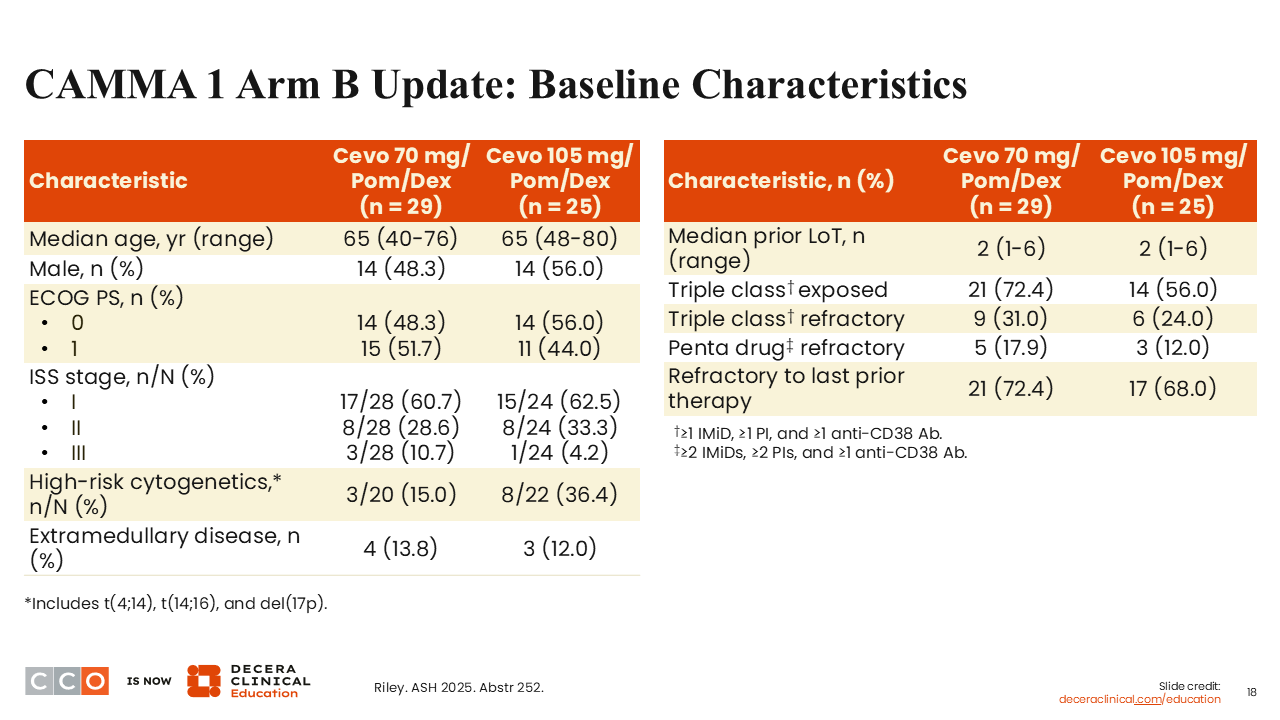
CAMMA 1 Arm B Update: Baseline Characteristics
Sagar Lonial, MD:
The baseline characteristics were relatively similar between the 2 cevostamab dosing groups.11
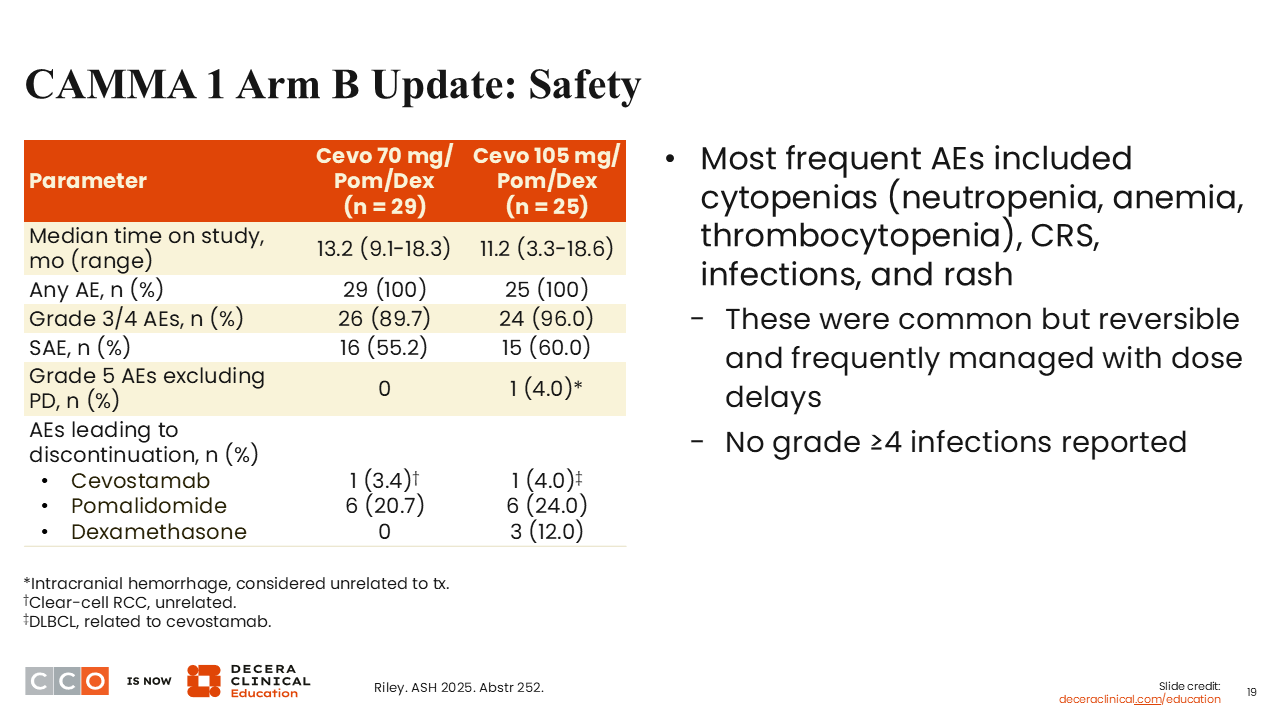
CAMMA 1 Arm B Update: Safety
Sagar Lonial, MD:
In terms of new AEs, there was not really anything appreciably different with the combination, except perhaps a little bit more hematologic toxicity when cevostamab is combined with pomalidomide, which is not a surprise.11
You certainly will see more neutropenia, anemia, and thrombocytopenia. CRS was not really increased once you started the pomalidomide after the step-up doses for cevostamab. I think that is a lesson learned that pretty much all bispecific antibody trials are doing now. You are getting through that initial step-up dose before you add in the immunomodulatory agent or the cereblon E3 ligase modulatory drug (CELMoD), depending on the specific trial.
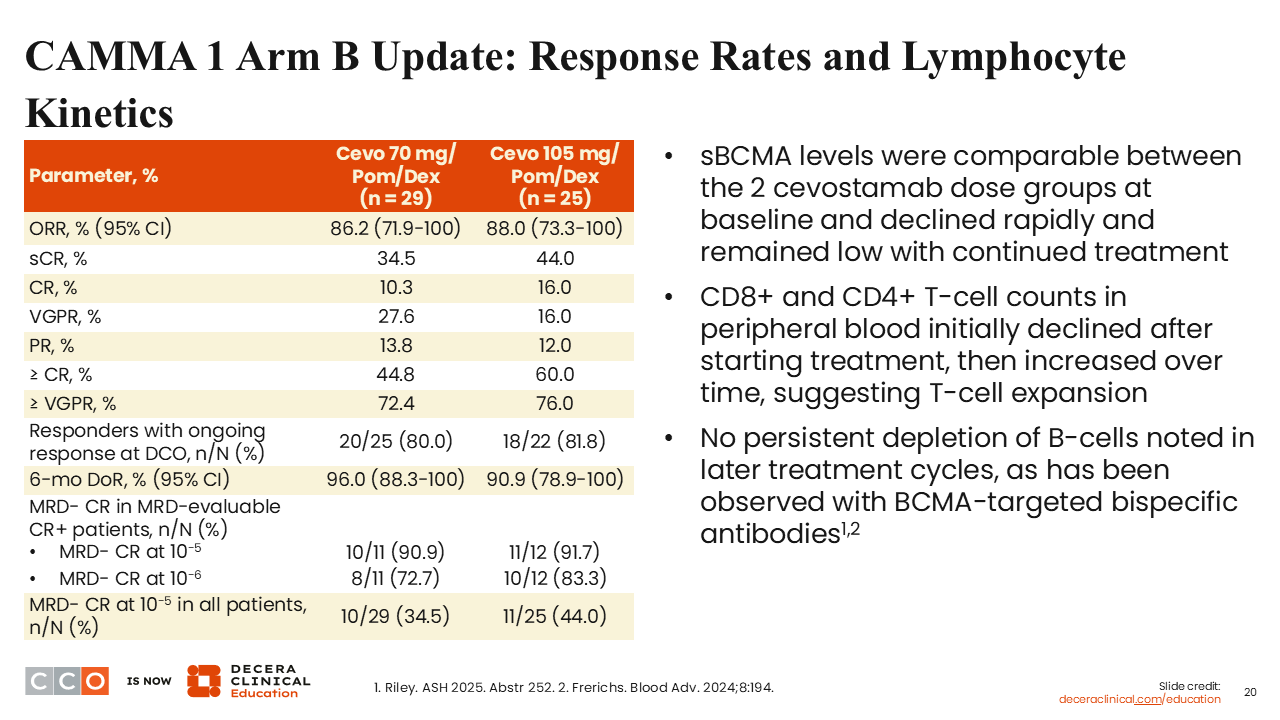
CAMMA 1 Arm B Update: Response Rates and Lymphocyte Kinetics
Sagar Lonial, MD:
The updated ORRs are 86.2% and 88.8% at the 2 dose levels, with 34.5% and 44.0%, respectively, achieving an sCR, and a very high fraction of patients with CR achieving MRD negativity—between 80% and 90%, depending upon whether you use 10-5 or 10-6 sensitivity of detection.11 Clearly, this is a highly effective treatment. With an 88% ORR, it is more effective than single-agent cevostamab has been shown to be in early trials,13 although those trials tended to be in more heavily pretreated patients, whereas this is a BCMA-naive patient population.
CAMMA 1 Arm B Update: Pharmacodynamic Effects
Sagar Lonial, MD:
The investigators looked at metrics to understand the impact of pomalidomide on T-cell activation as measured by soluble CD25. The addition of pomalidomide was associated with a reduction in T-regulatory cells. This has been shown to enhance the efficacy of bispecific antibodies and, in this case, enhances the efficacy of cevostamab.11,13
Conclusions
Sagar Lonial, MD:
In summary, the combination of cevostamab and pomalidomide/dexamethasone is clearly highly effective and appears safe. It does not add to the significant AEs associated with this concept of combining a bispecific antibody with a CELMoD or combining a bispecific antibody with an immunomodulatory drug (IMiD) that I think is moving across all bispecific antibodies.
I think it sets up a really interesting question. We have seen 2 sets of data now, combining bispecific antibodies with each other or with anti-CD38 antibodies.1,5 I think ultimately an interesting question for us as a field is: Are the CELMoDs or IMiDs better partners for the bispecific antibodies, or are other immune-based therapies like bispecific antibodies or CD38 antibodies the better targets for a combination strategy? Maybe we will end up doing it all. If you are talking about a doublet, is all-immune or immune with a CELMoD or an IMiD the better answer? I do not know the answer, but I think it would be a fun trial to try to put together to address some of those questions. These are really exciting data showing the activity of cevostamab plus pomalidomide.
Shaji Kumar, MD:
It is certainly interesting, especially in the context of us seeing more and more bispecific antibodies, especially a BCMA-targeted bispecific antibody used in combination with anti-CD38. Now, this gives an opportunity to use a similar strategy without any overlap, because you have a bispecific antibody with a different target, and, as you said, we might end up using all of them sequentially.
It will still be important ask whether the anti-CD38 should be sequenced first or with the IMiD first. Whether something else should happen in between is also an important question to be asked. Nevertheless, I think the data look strong. Of course, if we had a cohort of BCMA-exposed patients, that would have been data that we could have applied today, based on patients coming off the BCMA-targeted therapies that are becoming ubiquitous in this patient population.
Phase II Trial of Cevostamab Consolidation After Anti-BCMA CAR T-Cell Therapy in R/R MM
Shaji Kumar, MD:
We’ll next talk about the phase II STEM trial of cevostamab consolidation after anti-BCMA CAR T-cell therapy.16
CAR T-cell therapy has been quite paradigm-shifting in MM, especially in the relapsed setting, where it has moved all the way up to first relapse with cilta-cel.17 Ongoing studies are likely to make CAR T-cell therapy front-line treatment, at least for a fraction of patients.
The challenge that we see with CAR T-cell therapy, especially in patients with high-risk disease, is that the responses do not last as long as we would like.
Ongoing trials are looking at whether maintenance strategies—whether an IMiD or an anti-CD38 monoclonal antibody—could keep disease under control after patients receive CAR T-cell therapy and attain a deep response.
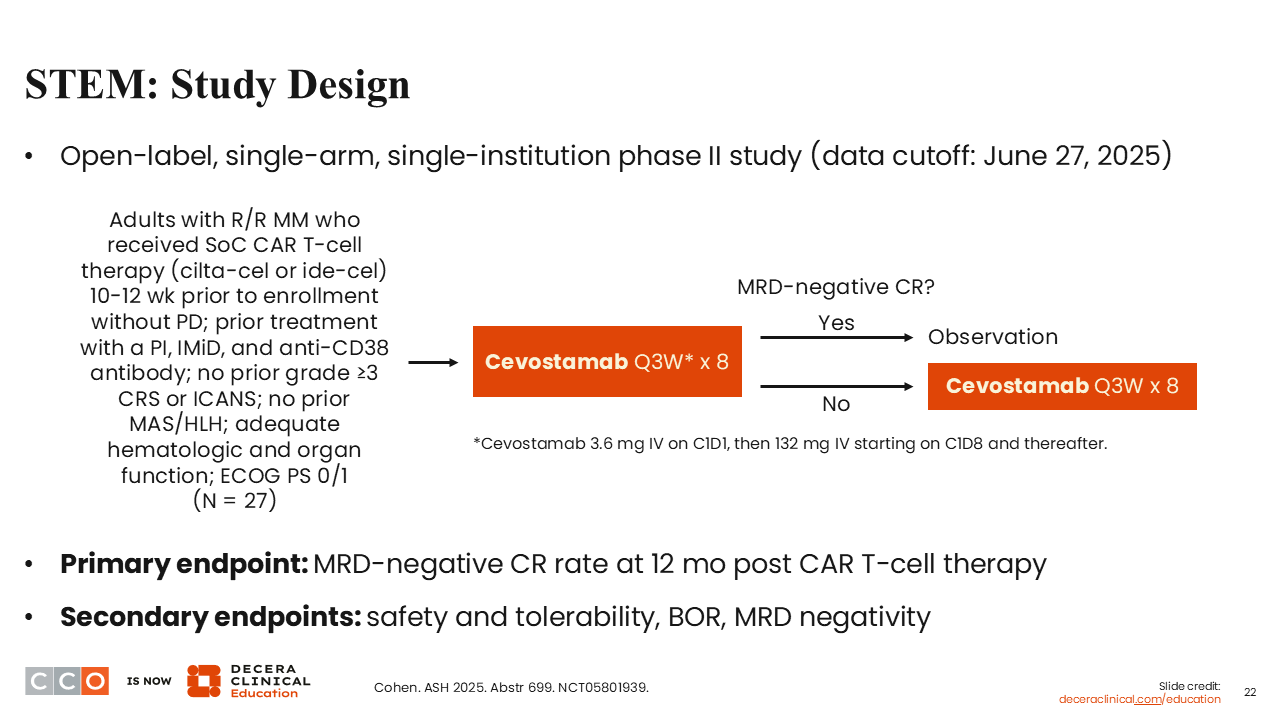
Many of those maintenance strategies, especially with IMiDs, have paralleled what we have done in the non–bispecific antibody or non–CAR T-cell setting, using continuous therapy until disease progression. However, this takes away 1 of the biggest advantages we have for CAR T-cell therapy, which is limited-duration therapy. Can we find something halfway between the 2 for those patients who still need some therapy after the CAR T to consolidate, where we do not need to give indefinite therapy? The STEM trial really tries to answer this question, to at least try to explore the hypothesis that we can give a limited-duration bispecific antibody (cevostamab) targeting a different antigen than the CAR T-cell to improve upon the durability of response.16
This open-label, single-arm phase II trial enrolled patients who had received a BCMA-targeted CAR T-cell, either cilta-cel or idecabtagene vicleucel (ide-cel), within the past 10-12 weeks. Patients could not have had a prior grade ≥3 CRS or immune effector cell–associated neurotoxicity syndrome (ICANS) or macrophage activation syndrome/hemophagocytic lymphohistiocytosis.16
Patients received cevostamab every 3 weeks for 8 doses. At that time, patients testing MRD negative would stop cevostamab, and those not MRD negative would receive an additional 8 doses and then stop. Either way, it is a limited-duration therapy with a maximum of 48 weeks of treatment with 16 doses.
The primary endpoint was the MRD-negative CR rate; again, this study was hypothesis-generating, so we want to try to get patients to a deep response. Eventually, we will get to learn about how long these responses last as well.
STEM: Baseline Characteristics
Shaji Kumar, MD:
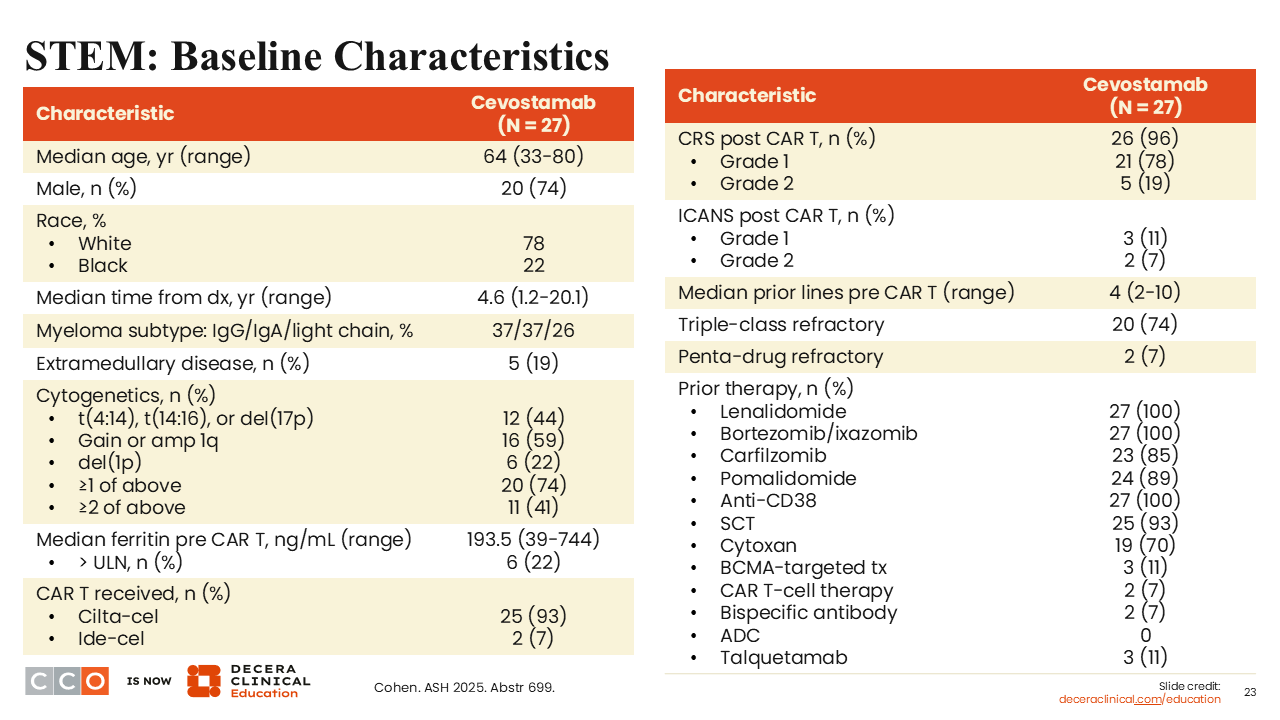
This was a single-institution study of 27 patients, so not a huge number. Patient characteristics were typical for patients undergoing a BCMA-based targeted CAR T-cell therapy.16 High-risk cytogenetic markers were present in at least half of patients; 74% were triple-class refractory. Patients had received a median of 4 prior lines of therapy, which included a variety of different treatments. All patients had received IMiDs and anti-CD38. Patients had received many of the proteasome inhibitors (PIs), and 93% had received a prior stem cell transplant.
STEM: Dosing Feasibility
Shaji Kumar, MD:
The study explored the feasibility of increasing doses.16 The dose that was eventually arrived at was a 3.6-mg 1-step-up dose, followed by 132 mg thereafter.

STEM: TEAEs
Shaji Kumar, MD:
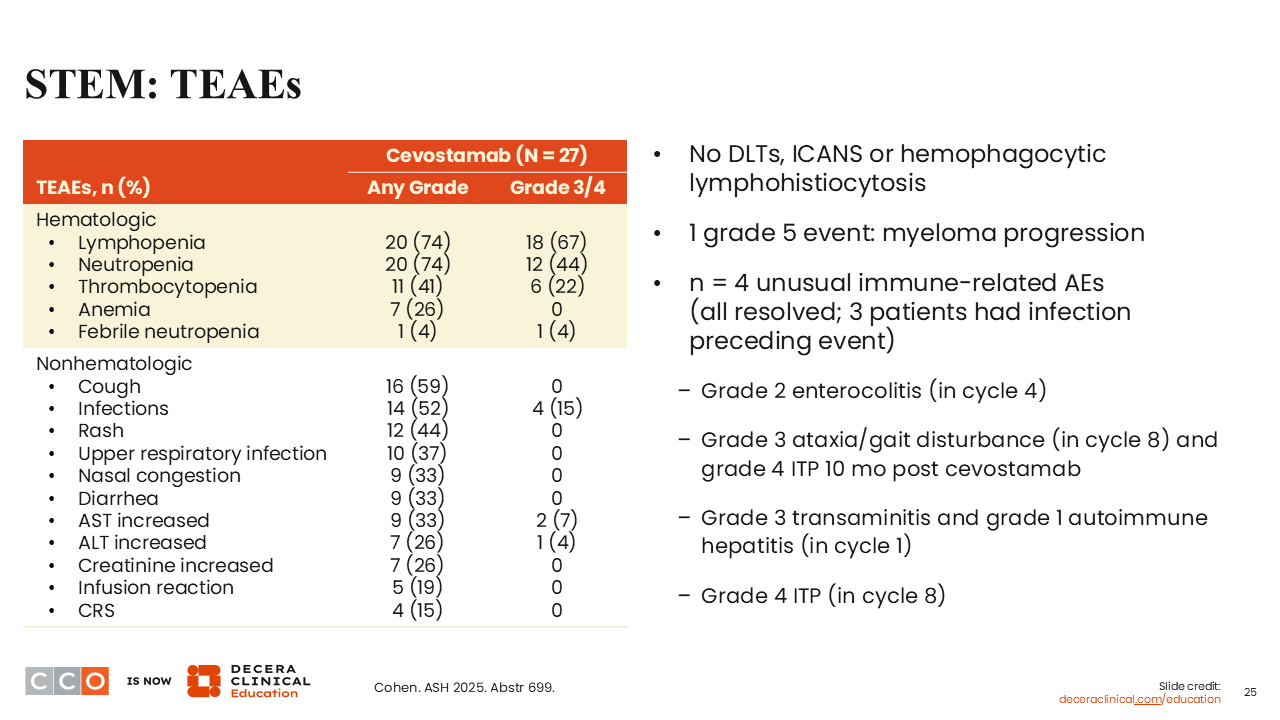
In terms of AEs, as one would expect from bispecific antibodies, there was hematologic toxicity in the vast majority of patients, including grade 3/4 lymphopenia in 67% and grade 3/4 neutropenia in 44%.16 Nonhematologic toxicities were mostly grade 1/2. There was a smattering of nonhematologic grade 3/4 toxicities, primarily infection (15%).
No cases of ICANS or hemophagocytic lymphohistiocytosis were reported. Unusual immune-related AEs occurred in 4 patients, which is something to think about. If this approach is studied in large groups of patients, this is something we need to keep an eye out for because it seems to stand out compared to what we see in other settings where we use bispecific antibodies. That may have something to do with the immune reconstitution that is already happening post CAR T-cell therapy.
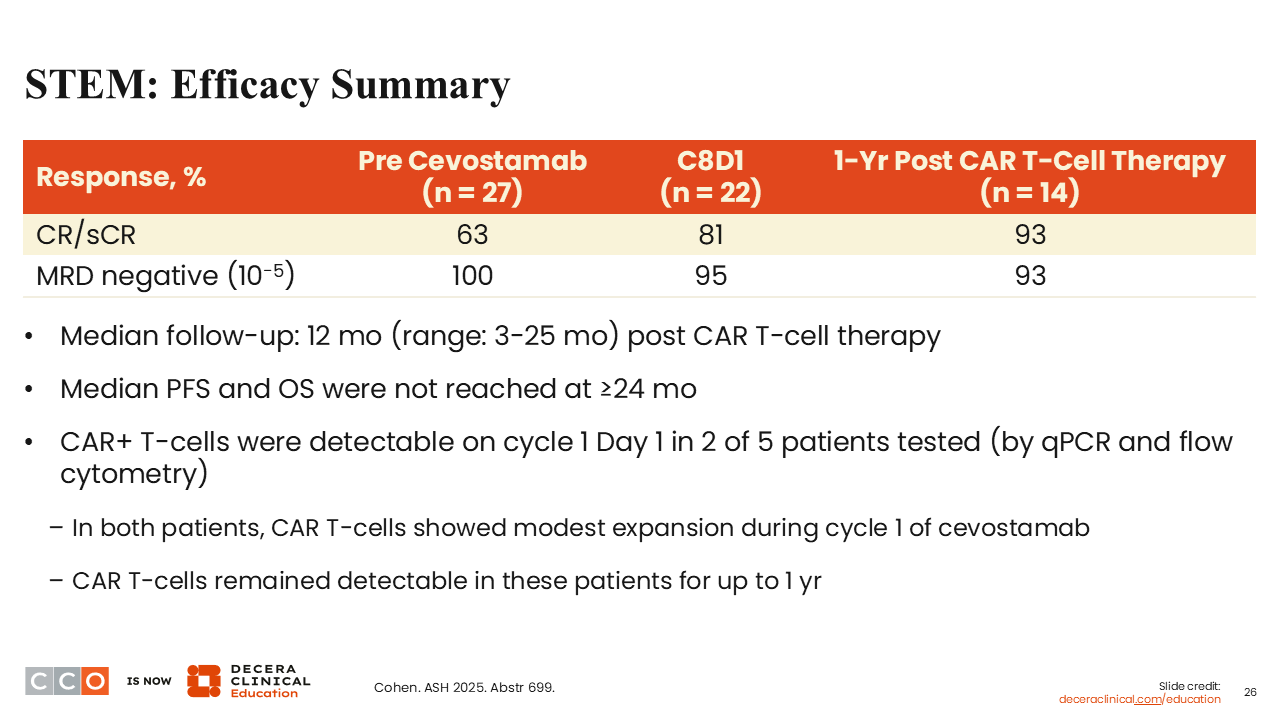
STEM: Efficacy Summary
Shaji Kumar, MD:
In terms of efficacy, of 14 evaluable patients, 93% attained MRD negativity (10-5) at 1-year post CAR T-cell therapy.16 One thing to remember is that the vast majority of these patients are already MRD-negative. That is 1 of the disadvantages of really getting a good handle on what this means for the future. We do not have any data with MRD negativity at a deeper level in this particular study.
Median PFS was not reached at 2 years. Of interest, CAR T-cells still remained detectable for up to 1 year in 2 of 5 patients tested, highlighting that the use of bispecific antibodies did not eliminate the CAR T-cells, which certainly is something one might worry about in this setting.
Conclusions
Shaji Kumar, MD:
In conclusion, this looks more like a feasibility study to me. I think we are all looking at ways to make the CAR T-cell responses last longer, and I look at this as a time-limited approach that can achieve the same thing, without a significant addition of toxicity for these patients.
Sagar Lonial, MD:
I think trials like this represent some of the most exciting things that we are doing with so many immune targets in myeloma. We are now exploring limited-duration therapy across the board and doing so by switching targets.
The rationale behind this is really solid. We know cells that end up relapsing after CAR T therapy are often BCMA low.18 The idea of coming in by switching the target afterwards makes a lot of sense. FcRH5 is a target that is expressed on some of the more primitive myeloma cells. Perhaps you are getting closer to that stem cell concept by using cevostamab after a BCMA-directed approach.
This concept of switch-target limited-duration therapy is exactly where I think we are going to end up in the future, in more of an ALL-like treatment approach for myeloma, as opposed to a treatment until progression and putting 10 different drugs together at 1 time. This is more of a sequential approach, trying to chip away at whatever may be left. I think you are right that it is hard to know what is left, given that they were MRD-negative going into the trial, or at least a lot of them were.
We know that a majority of patients who are MRD-negative will relapse anyway. I think getting an idea of PFS will be a reasonable endpoint to evaluate whether this strategy makes sense over time.
The other point I would make is about the atypical immune-related events. We are seeing a lot of that now, after cilta-cel, for instance. How much of that was related to giving a bispecific antibody after a CAR T vs just what you would have seen anyway? I have a bunch of patients with an odd hand arthritis that is treated as an autoimmune disorder. We are seeing some of the thyroid immune AEs that have been seen before as well. I think that in a small study, it is hard to know what it means, but it may not necessarily be an additive effect. It may have been the result of the CAR T therapy itself.
Extended Efficacy and Safety Outcomes With Cilta-cel in Patients With R/R MM With Standard-Risk Cytogenetics
Sagar Lonial, MD:
Let’s switch gears and review this longer-term follow-up from the CARTITUDE-4 study, focused on cilta-cel in patients with standard-risk genetics.19
In the CARTITUDE-1 trial, approximately a third of patients with heavily pretreated R/R MM were in an ongoing CR at 5 years.20 CARTITUDE-4 enrolled patients with 1-3 prior lines of therapy and led to the FDA approval of cilta-cel in first relapse.21 In that study, cilta-cel demonstrated a benefit in functionally high-risk patients, and I think that is where many of us are using cilta-cel in the context of first-relapsed myeloma.22
This analysis was an attempt to evaluate the relative benefit of cilta-cel in patients with standard-risk cytogenetics.19 We are continuing to talk about the best treatment approach for a standard-risk patient, given the wealth of different treatment options we have, including MajesTEC-3,1 which we discussed above.
CARTITUDE-4 evaluated cilta-cel vs SoC therapy consisting of physician's choice of PVd or DPd given until progression.19 The primary endpoint was PFS, which was met a long time ago, demonstrating superiority of the CAR T-cell therapy over SoC.21
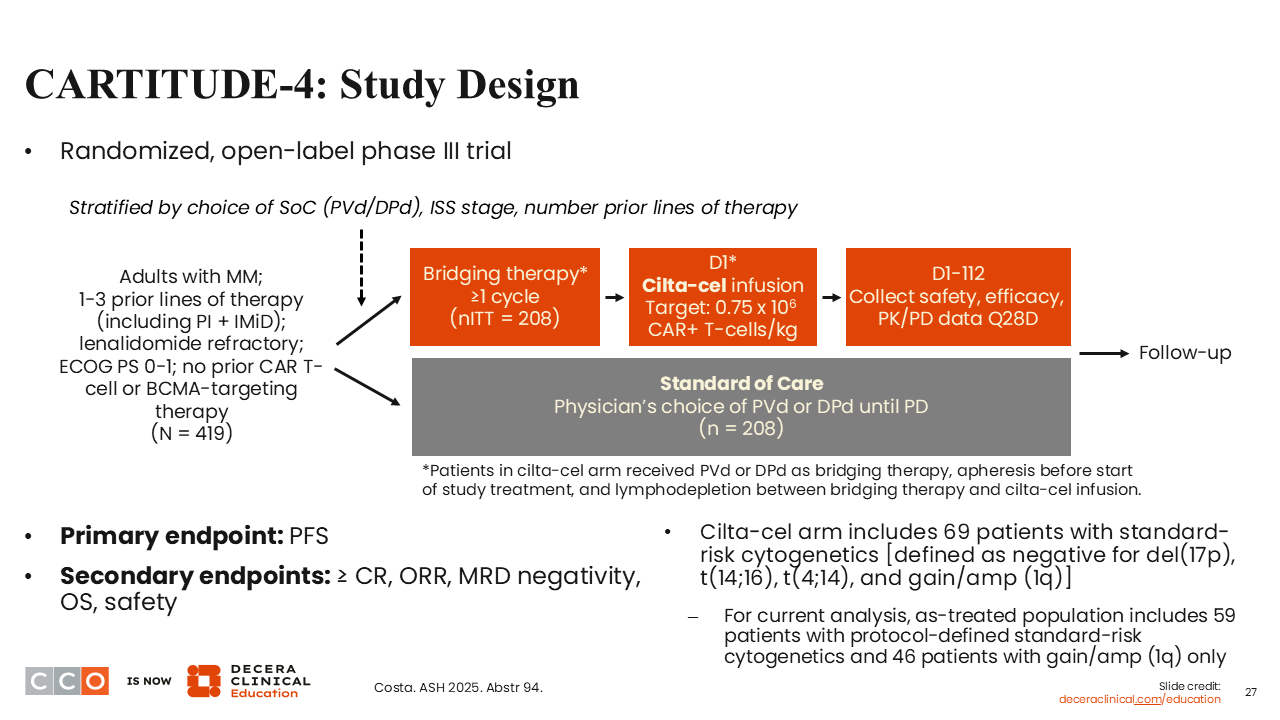
Cilta-cel for Standard-Risk MM: Baseline Characteristics
Sagar Lonial, MD:
These are the baseline characteristics of patients with standard-risk cytogenetics in CARTITUDE-4 and those who had a gain/amplification (1q), compared with patients with standard-risk cytogenetics in CARTITUDE-1, which was a later-line trial.19 The groups were similar in terms of resistance states, number of prior lines, etc, with the exception that CARTITUDE-1 is a much later line trial overall.
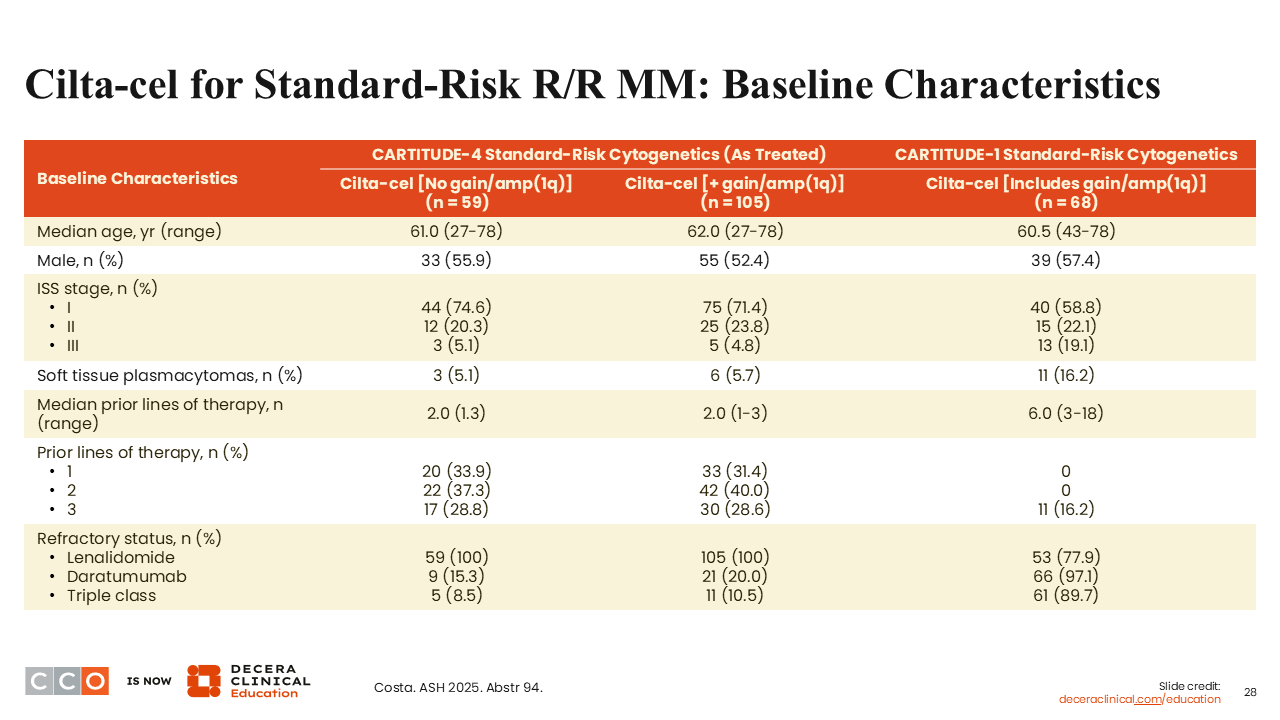
Cilta-cel for Standard-Risk R/R MM: Response
Sagar Lonial, MD:
Response rates were very high across the board. The ORR in CARTITUDE-4 was 100%, with rates of CR or better of 90% to 92%.19 A large proportion of patients achieved CR or very good partial response or better, both in patients with and without a gain/amplification (1q).
Cilta-cel for Standard-Risk R/R MM: PFS and OS
Sagar Lonial, MD:
What is really quite impressive is the 30-month PFS rates of 80.5% in patients without gain/amp(1q).19
Remember, in MajesTEC-3, we saw a 3-year PFS rate of 83% for all patients, and yet we are at a 30-month PFS rate of 80% in standard-risk patients.1,19 Also, the 71% 30-month PFS rate in patients with gain/amplification (1q) is pretty good for high-risk, or at least not standard-risk, patients, which is probably how I would describe this group. These may not all represent high-risk patients, but certainly they are not standard risk as defined by genetics alone.
Again, this is significantly better than what we saw in CARTITUDE-1 for a similar patient population.

Cilta-cel for Standard-Risk R/R MM: Survival in Patients Alive and Progression Free at 1 Yr
Sagar Lonial, MD:
Among patients with standard-risk cytogenetics (no gain/amplification [1p]) with an early sustained response to cilta-cel, 93.1% were alive and progression-free at 30 months.19 Certainly, this is very respectable data.

Cilta-cel for Standard-Risk R/R MM: Safety
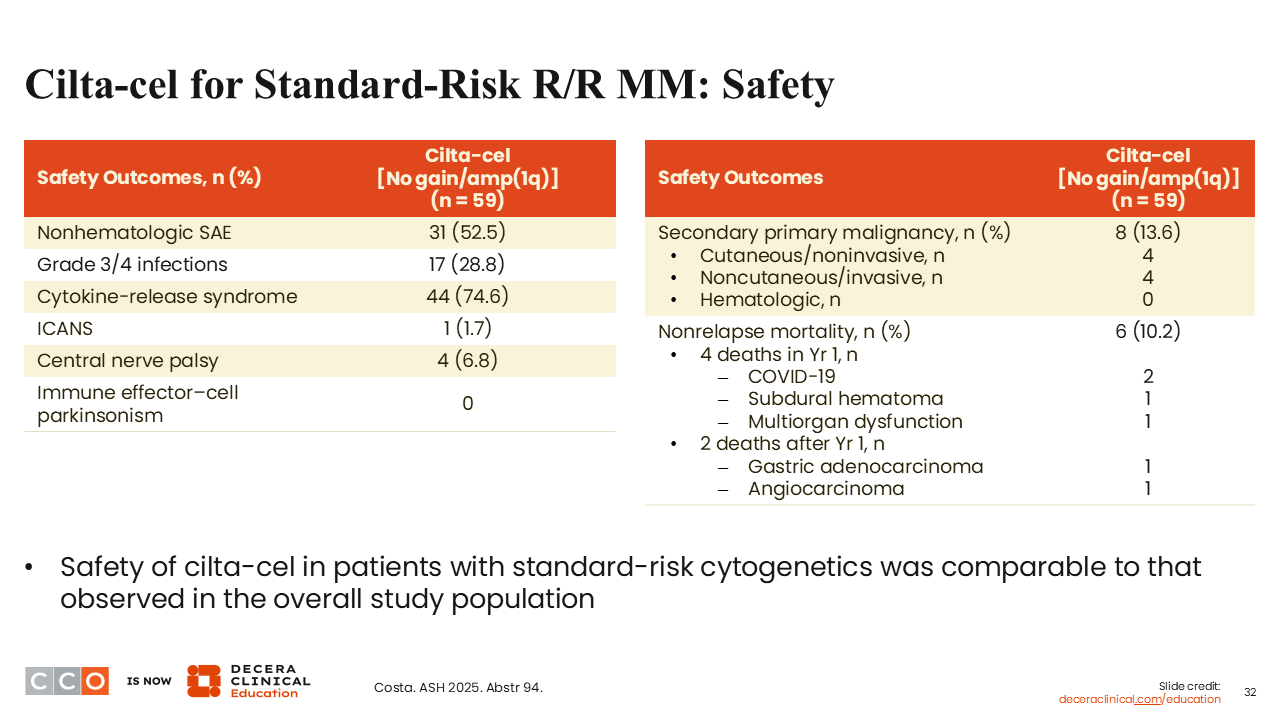
Sagar Lonial, MD:
Safety was not appreciably different between the groups; we see the same AEs that we expect with cilta-cel overall.19 Only approximately 4% of patients had any central nerve palsy, and no patients had parkinsonism.
Conclusions
Sagar Lonial, MD:
I think this almost sets the stage between the MajesTEC-3 regimen vs cilta-cel for standard-risk early relapsed MM. This has significantly raised the bar we typically have considered for median PFS from 36 months, being about as good as you can get in this space. Here, we are talking about a PFS rate of 80% with cilta-cel at 30 months to 83% at 3 years with Tec/Dara.1,19
Although the debate on bispecific antibody vs CAR T-cell therapy will continue, remember the relative advantage of CAR T therapy is that once you are done with therapy, you are done with therapy. You are going to be under pretty significant supportive care for the next 6-12 months. Certainly, the risk of treatment-related AEs drops as you get further away from the CAR T infusion. I think it is really a great position to be in. It raises the question about early CAR T-cell therapy for standard-risk myeloma.
Shaji Kumar, MD:
It is really interesting to look at the standard-risk patients, because most analyses focus on the high-risk subgroup. If you look purely from /he standpoint of what is the best option for standard-risk patients in first relapse, these data look pretty good, and comparable to the Tec/Dara data. However, I don’t think it necessarily answers the question of which way to go for those patients. It is certainly something worth exploring for the future.
I don’t know if we can necessarily use these data to say that we have to use CAR T in the first relapse vs later on. The CARTITUDE-1 vs CARTITUDE-4 comparison does not really contribute much to that conclusion, because we are talking about patients with a median of 6 prior lines of therapy with CARTITUDE-1. We know patients in later lines of therapy are always going to get a lower durability of response with any therapy that we apply for them. That aside, just in absolute terms, I think the PFS at 2.5 years is pretty good compared to what we had in the past.
iMMagine-1 Update: Phase II Study of D-Domain BCMA-Directed CAR T-Cell Therapy, Anitocabtagene Autoleucel, in R/R MM
Shaji Kumar, MD:
Current CAR T-cell therapies have high efficacy but also come with definite toxicity, some of which can be devastating, particularly neurological toxicity. This can make the argument for using CAR T-cell therapy in the early relapse setting a little bit weaker than we would like. I think that is where some of the new CAR T-cells are probably going to make an impact in the field. Let’s discuss the updated data from the iMMagine-1 study evaluating anitocabtagene autoleucel (anito-cel) in patients with R/R MM.23
Anito-cel is a novel CAR T-cell therapy that targets BCMA but has clear differences from current CAR T therapies in terms of its structure. Anito-cel uses a novel D-domain binder that has a fast off-rate and has a high transduction efficiency.24 This could lead to potent tumor killing without the sustained inflammation and some of the consequent AEs we see with current CAR T-cell therapies, like CRS and neurologic toxicity. We have previously seen the initial data from this phase II trial, showing that anito-cel is highly effective.25 We now have updated data from the entire cohort of patients.23
The iMMagine-1 study enrolled patients with R/R MM with 3 or more prior lines of therapy who had been exposed to an IMiD, a PI, and an anti-CD38 antibody. A total of 118 patients underwent lymphodepletion, and 117 received therapy.23
iMMagine-1: Baseline Characteristics
Shaji Kumar, MD:
The baseline characteristics were as one would expect for patients in later lines of therapy. Patients had received a median of 3 prior lines of therapy and most (87%) were triple class-refractory.23 EMD was present in 18% of patients. Many patients had significant tumor burden, with 35% having more than 30% plasma cells in the bone marrow; 40% had high-risk cytogenetics.
iMMagine-1: ORR, Depth of Response, and Survival
Shaji Kumar, MD:
The ORR was very high at 96%, with 74% of patients attaining an sCR or CR.23 The 2-year PFS rate was 61.7%. We anticipate that the median PFS in this study will probably be between 2 and 3 years, which again is quite comparable to what we have seen with cilta-cel. The 2-year OS rate was 83%.
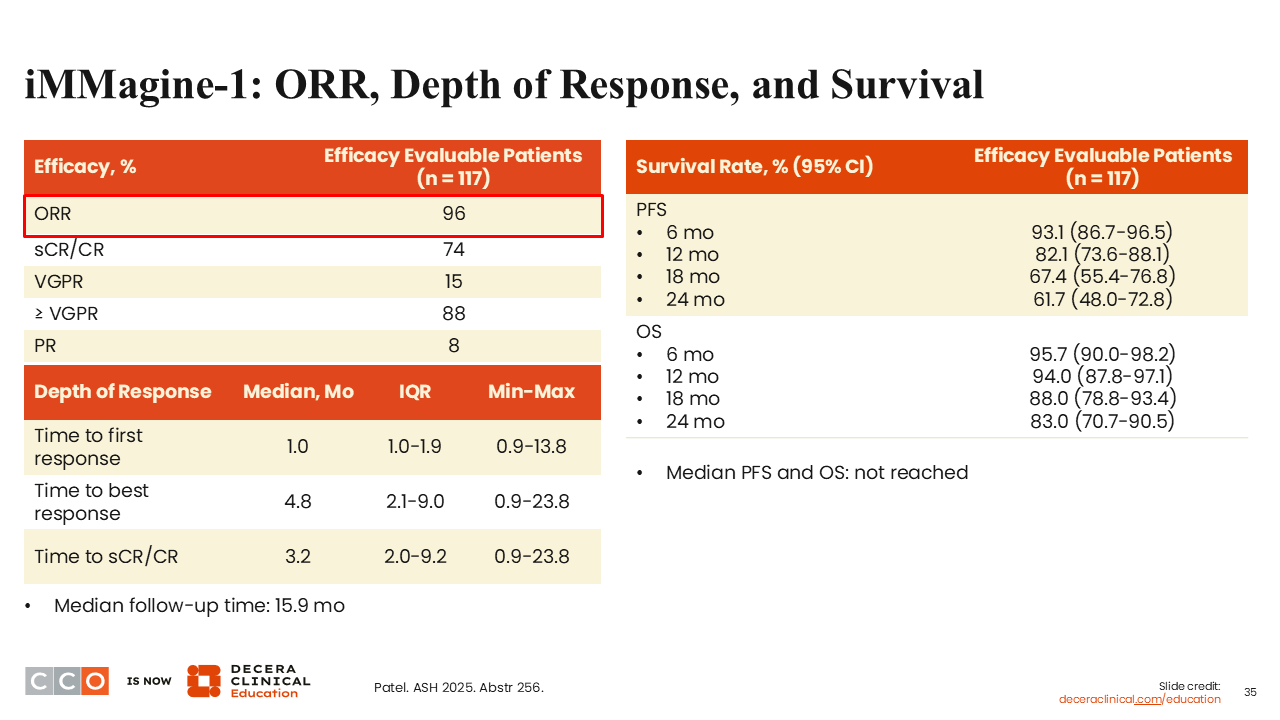
iMMagine-1: MRD Status
Shaji Kumar, MD:
The MRD negativity rate was again very high rate, with 95% of evaluable patients attaining MRD negativity at 10-5.23 Clearly, the responses that we are getting from anito-cel appear to be quite deep.

iMMagine-1: Safety
Shaji Kumar, MD:
The toxicity profile was very similar to what we have seen with other CAR Ts, with the notable exception that there was no delayed or non-ICANS neurotoxicity, parkinsonism, cranial nerve palsies, Guillain-Barré syndrome, or immune effector cell–associated enterocolitis in this patient population.23
Granted, the follow-up is a little shorter than what we have seen with the cilta-cel long-term studies. Nevertheless, the conspicuous absence of neurologic toxicity, especially delayed neurologic toxicity, appears to be quite striking. Moreover, a majority of patients (95%) had no CRS or CRS that was resolved within 10 days, so less of a burden than what we have seen with current products.
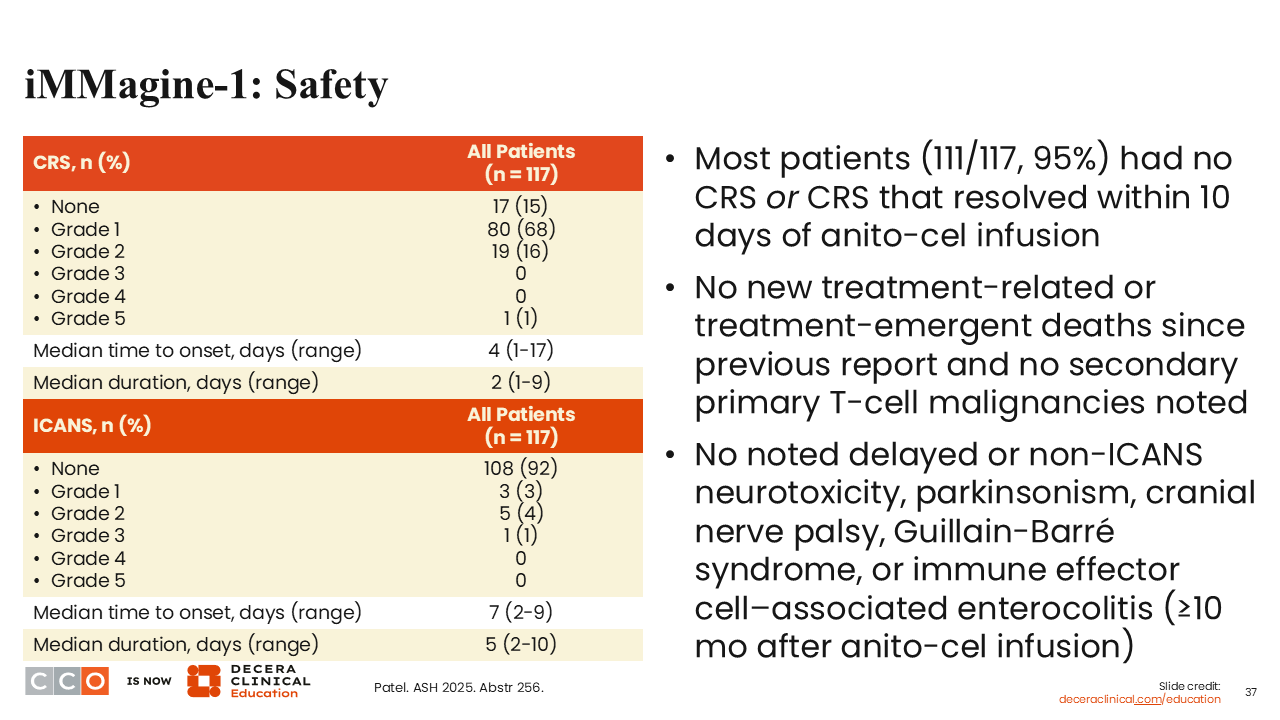
iMMagine-1: Safety Continued
Shaji Kumar, MD:
Looking at other toxicities, you do see infections in approximately a third of patients, some of which are COVID.23 Hematologic toxicities were very similar to what we have seen with current CAR T-cell therapies. Approximately half of the patients (43%) developed hypogammaglobulinemia.
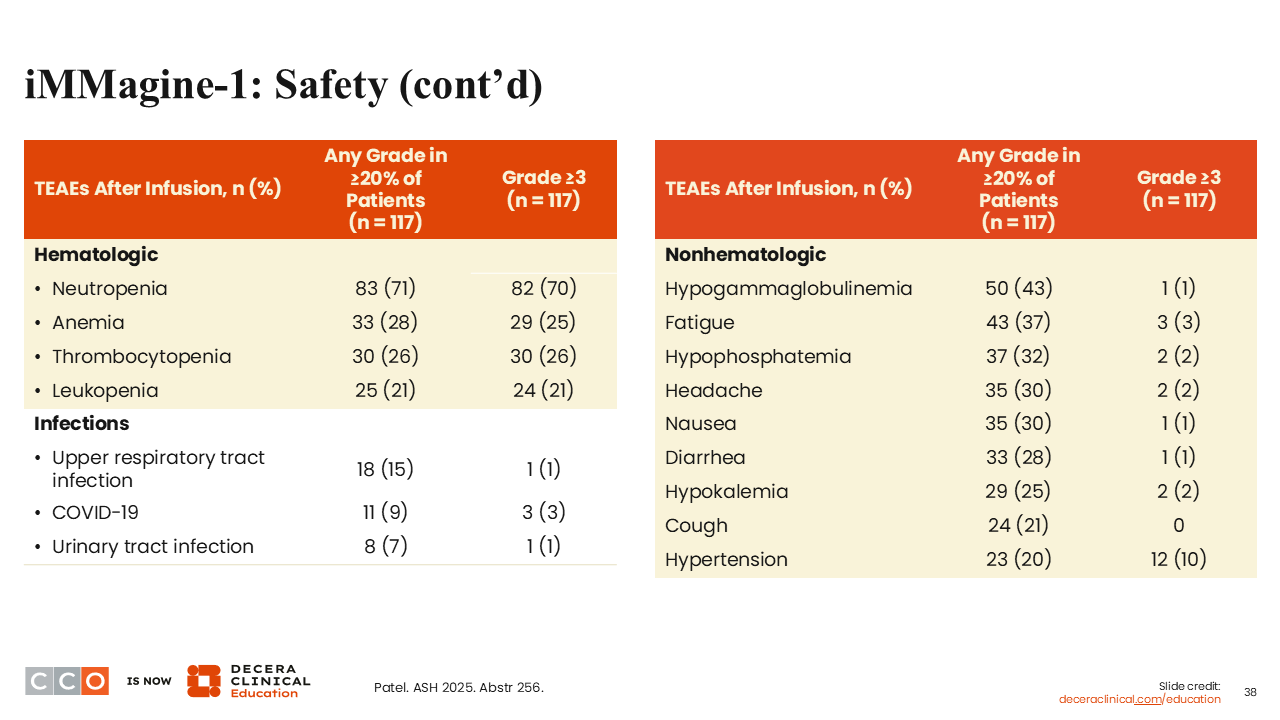
Conclusions
Shaji Kumar, MD:
Although anito-cel would be the third BCMA-directed CAR T-cell therapy to become available if it is approved, it certainly appears to have some novel characteristics. In particular, the degree of CRS appears to be lower, but more of more importance, there appears to be a lack of delayed neurotoxicity, at least so far. This may all be attributed to the novel D-domain binder with anito-cel. Certainly, longer-term follow-up is needed. Moreover, this more favorable safety profile seems to be happening without any compromise in efficacy. We might be getting closer to the sweet spot where we have the efficacy we want without the toxicity we do not want.
Sagar Lonial, MD:
We have seen that different products can have very different characteristics. We see differences in AEs and in efficacy with ide-cel vs cilta-cel.26 Certainly, in a small phase II study, the efficacy data here look quite good, and the toxicity data look relatively minimal compared to other CAR T-cell therapies.
With a note of some level of caution, I will say that with cilta-cel, we did not see many of the toxicities in large numbers or percentages until it was FDA-approved and we started treating a lot more patients. For example, enterocolitis was not something that we fully appreciated until we started treating a lot more patients later on. The cranial neuropathies were there, so not seeing that is certainly very exciting. I would just like to see a larger number of patients treated before we give the moniker that anito-cel does not have the neurologic toxicity that we saw with cilta-cel or even to a lesser extent with ide-cel.
DURGA-1: Phase Ib/II Study of AZD0120, a BCMA/CD19 Dual-Targeting CAR T-Cell Therapy, in R/R MM
Sagar Lonial, MD:
The final abstract we’ll discuss is the DURGA-1 study of the BCMA/CD19 dual-targeting CAR T-cell therapy AZD0120 in R/R MM.27
On that theme of trying to continue to improve upon the efficacy of CAR T-cells, 1 of the challenges that we struggle with is the manufacturing time. Often, you can get a patient to apheresis, but controlling disease in that time between apheresis and infusion can be a real challenge, particularly in patients in later lines of therapy.
The DURGA-1 trial sought to do 2 things: first, to evaluate the relative benefit of adding a second target, in this case CD19, to BCMA-targeted CAR T-cell therapy. There are some theoretical ideas about perhaps hitting an earlier progenitor, as we talked about with cevostamab, that may be able to eliminate some of the "stem cells." That hypothesis has been around for quite a while now.
The second is perhaps if you manufacture quicker, in this case, in less than 3 days, you may not need bridging therapy. You can apheresis a patient and then know that within a week you are likely going to start chemotherapy and get them a CAR T-cell product. Sooner is definitely better than later. These 2 things really represent to me the potential advantages of the product evaluated in this trial.
This was a phase Ib study looking at a relatively small number of patients so far, trying to establish the recommended phase II dose with standard apheresis and lymphodepletion.27 Two dose levels were evaluated, 1 x 105 cells/kg and 3 x 105 cells/kg, looking at efficacy, safety, and pharmacodynamics.
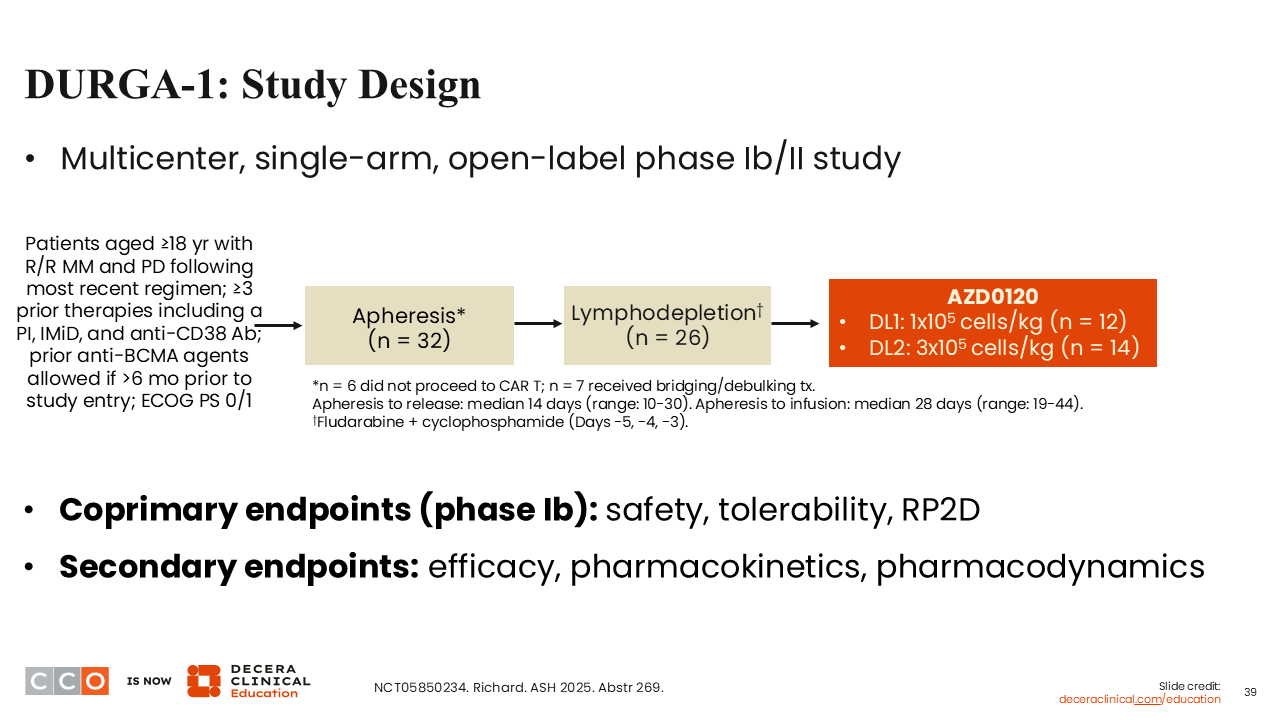
DURGA-1: Baseline Characteristics
Sagar Lonial, MD:
Patients had received a median of 4 prior lines of therapy, so this was a little bit earlier than what we saw with KarMMa-1 or CARTITUDE-1, which both enrolled patients with a median of 6 prior lines of therapy.27,29,30 I think that this reflects current practice, where patients are going to get CAR T-cell therapy earlier in the course of their disease. A few patients (23%) had seen a prior BCMA-directed therapy, but the majority had not.
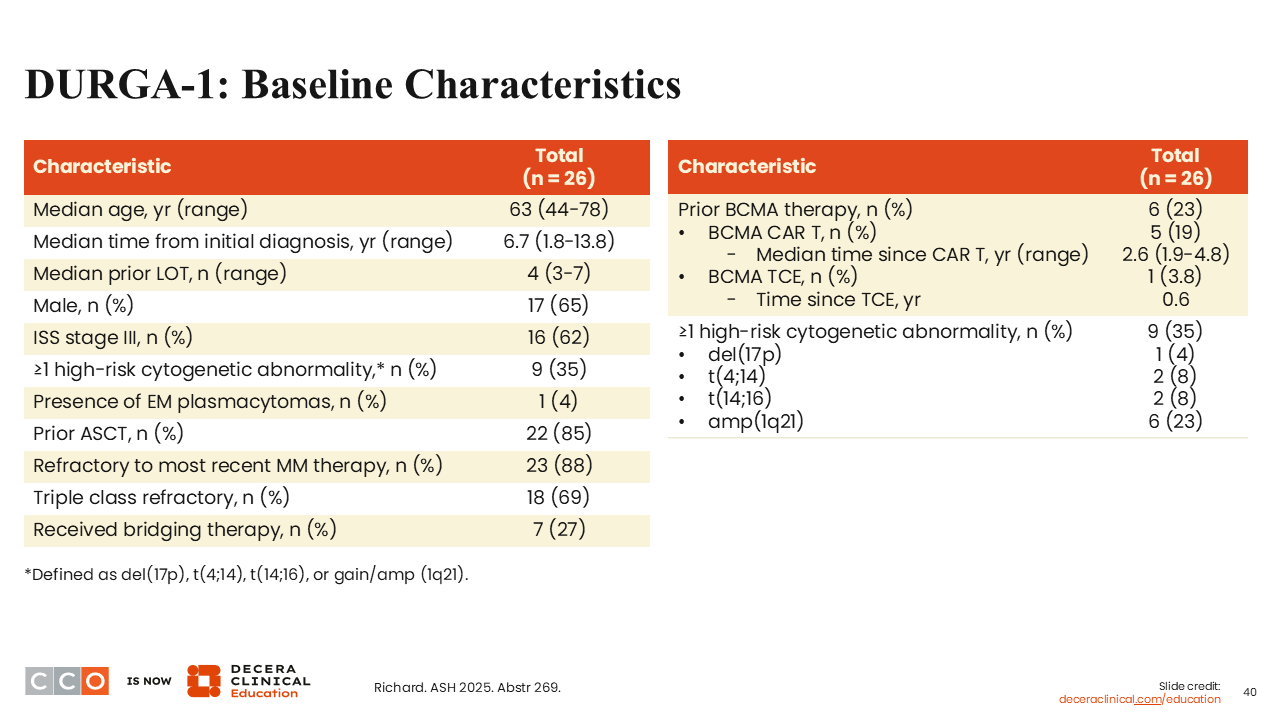
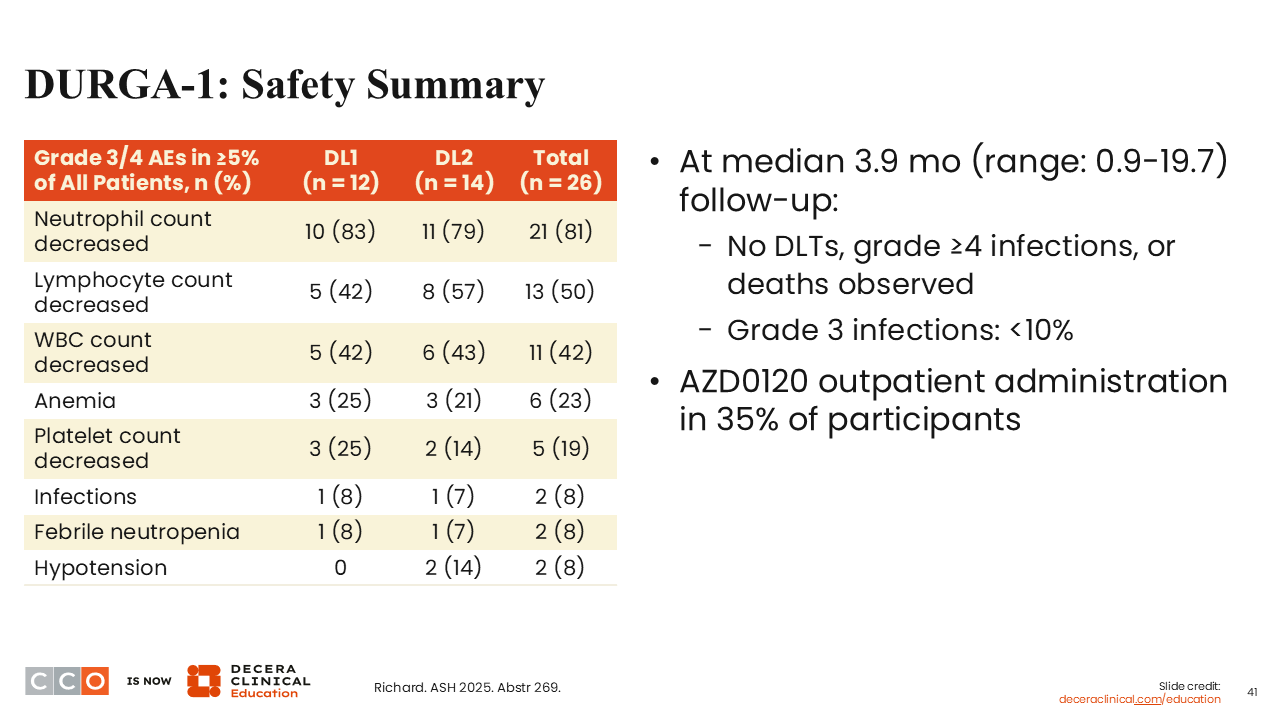
DURGA-1: Safety Summary
Sagar Lonial, MD:
Looking at the safety summary, the hematologic toxicity is not a surprise. Patients are getting lymphodepletion, which often causes neutropenia, anemia, and thrombocytopenia. There were no dose-limiting toxicities seen early on, after a median follow-up of only 3.9 months.27 Infections and neutropenia were as one would expect with this therapy approach.
Of interest, the CAR T-cell therapy was administered as an outpatient in 35% of patients.
DURGA-1: CRS and ICANS
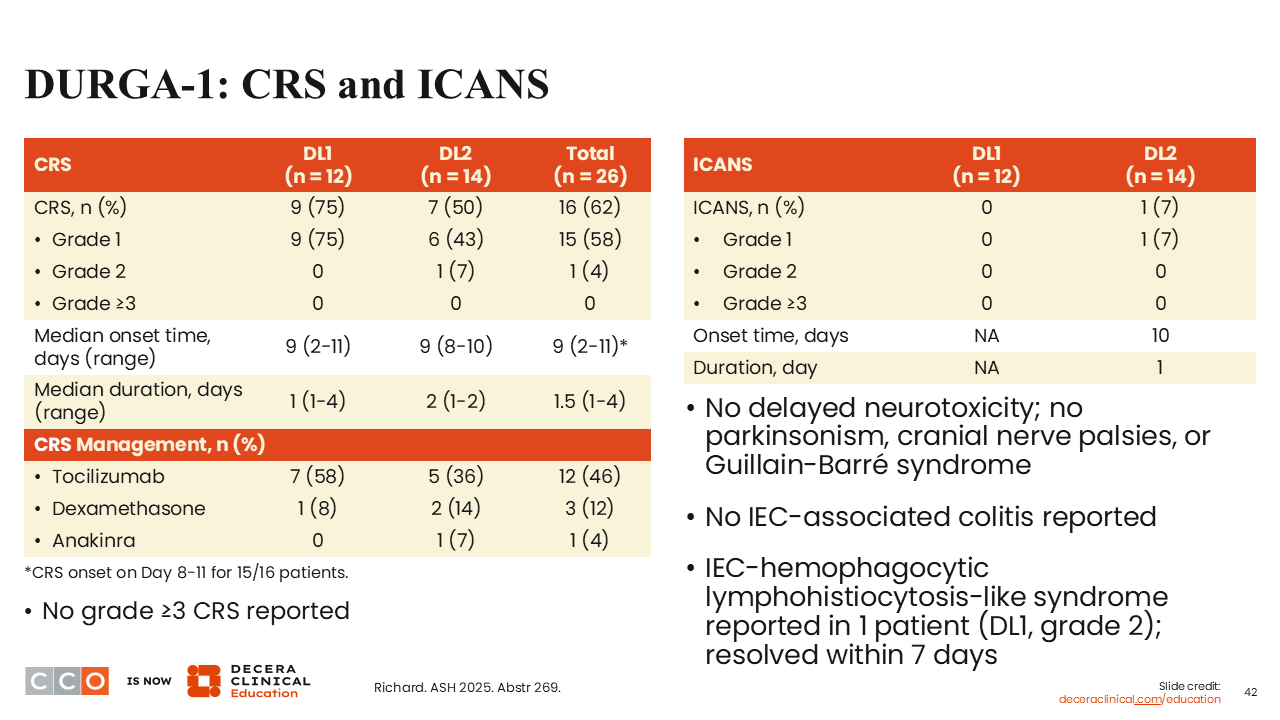
Sagar Lonial, MD:
CRS and ICANS outcomes are shown here.27 There was some CRS and 1 grade 1 ICANS event that lasted for 1 day. As of yet, with this short follow-up and with a relatively small number of patients, there has been no delayed neurotoxicity, parkinsonism, cranial neuropathies, Guillain-Barré syndrome, or enterocolitis. One patient had a grade 2 hemophagocytic lymphohistiocytosis–like syndrome that resolved within 7 days.
DURGA-1: Efficacy, MRD Status, and Cellular Kinetics
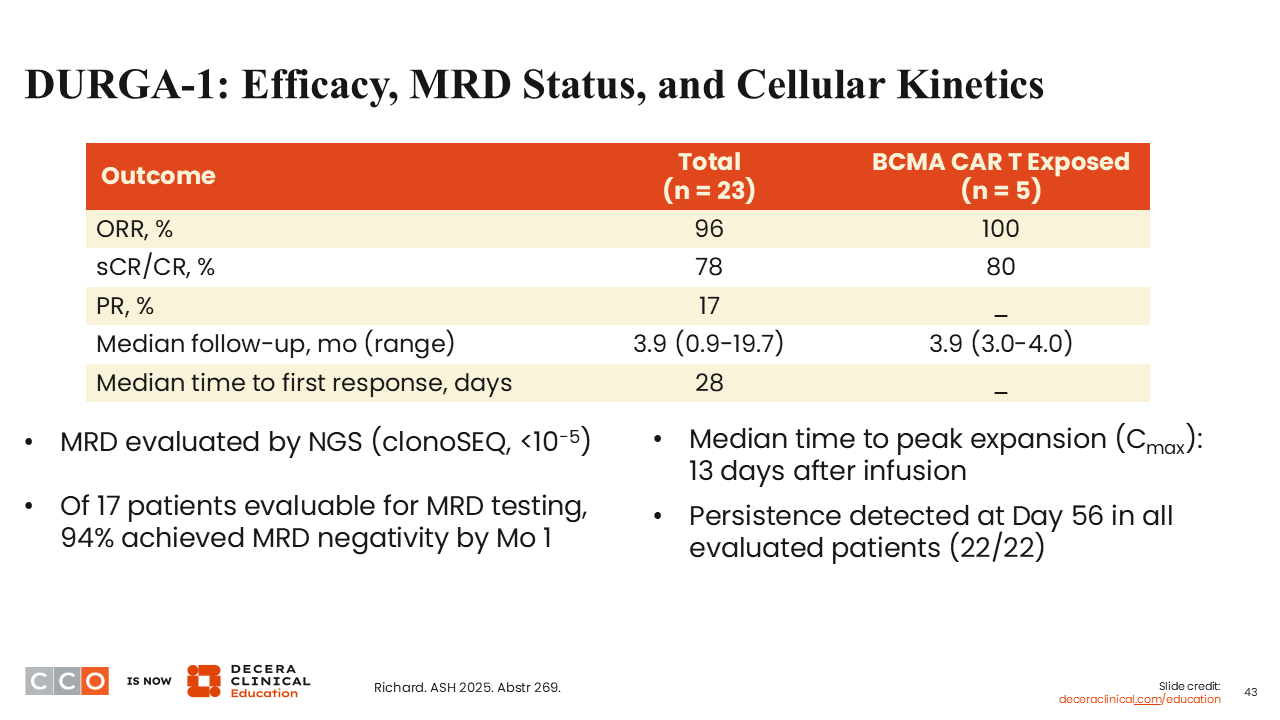
Looking at efficacy, even though this is a dose escalation cohort, 78% of patients achieved an sCR/CR, and of the 17 patients evaluable for MRD testing, 94% achieved MRD negativity by Month 1.27
We are always skeptical about Month 1 MRD data, as Month 3 is probably more meaningful because the marrow has truly recovered after lymphodepletion. All of the cytokine storm is gone within the first few weeks, but certainly 94% is a reasonable number and is encouraging overall. Of interest, they did see persistence at Day 56 in all 22 patients evaluated, which appears to be longer than we have seen with other CAR T products.
Conclusions
Sagar Lonial, MD:
In summary, this abstract reported encouraging data. It seems like there may be less neurologic toxicity, but it is very early. I think the rapid manufacturing time is certainly very encouraging, and there was nothing untoward or unexpected with this process. The MRD negativity data is very encouraging. In terms of dual targeting, although CD19 may not be my first choice as a second target if you are going to do a dual targeting CAR T, this certainly suggests as proof of principle that it can be done. We are participating in this trial and looking forward to putting some patients on to see what it looks like in a larger patient population.
Shaji Kumar, MD:
I think the data looks quite interesting, especially given the background of this particular CAR T, with multiple trials having been done in China.31,32 We have seen some longer-term results with AZD120 in newly diagnosed patients, where the responses appear to be quite durable, and almost all patients respond with deep MRD-negative responses.31
Regarding the concept of targeting 2 different antigens, even though CD19 might seem counterintuitive for a plasma cell population, there has always been the argument of the stem cell-like precursor stage that might be targeted through this strategy. Irrespective of whether that is true, we are certainly seeing some very good data with this approach. Again, CAR T is moving towards not only the goal of lower toxicity and high efficacy, but also almost off the shelf, where by the time we can get the patients ready for the CAR T therapy, you already have the product back in hand.