CME
Preparing for the Future: Integration of Second-line Therapies After Hydroxyurea for PV and ET
Physicians: Maximum of 0.25 AMA PRA Category 1 Credit™
Released: April 29, 2026
Expiration: October 28, 2026

Activity
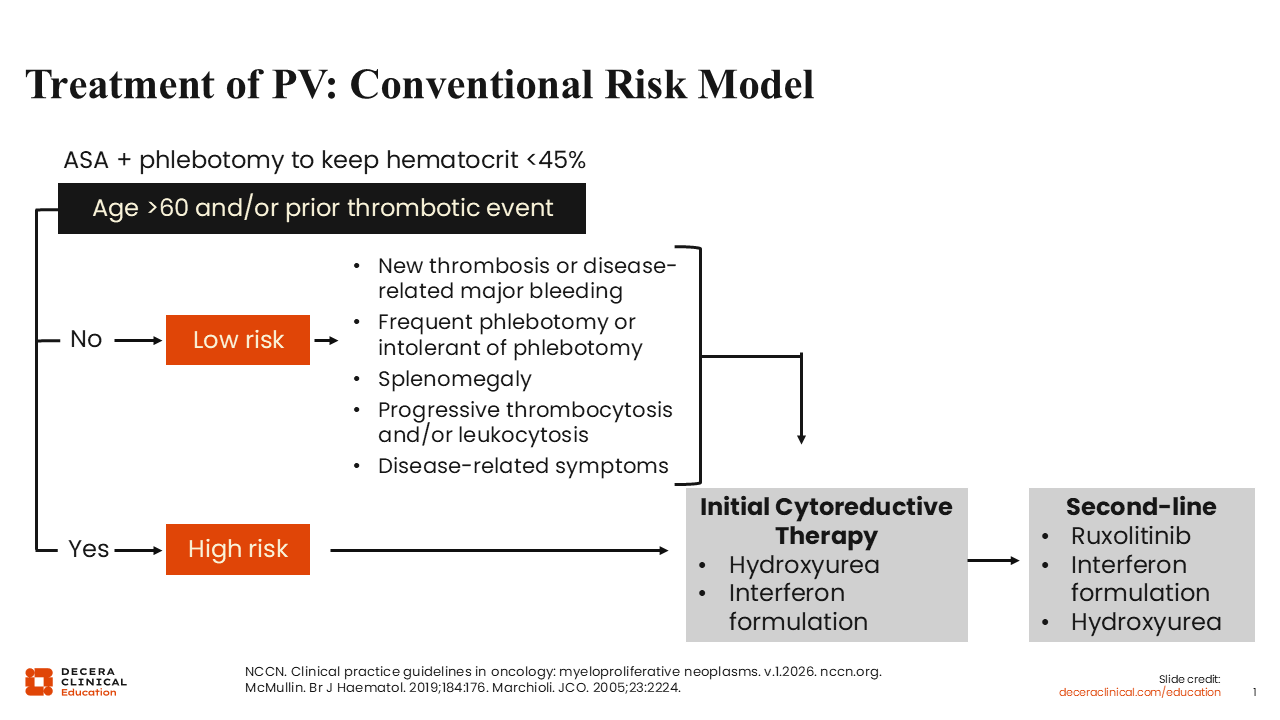
Treatment of PV: Conventional Risk Model
The conventional risk model has long been used to guide PV treatment. This risk model takes into account patient age, which is subdivided into older than 60 years or 60 years or younger, and any history of either a venous or thrombotic event.1-3
If a patient is either aged older than 60 years or has a history of a thrombotic event, he or she has high-risk PV. The absence of both constitutes low-risk PV. For both low-risk and high-risk PV, aspirin is recommended unless there is a contraindication. Phlebotomies are also recommended to keep the hematocrit <45% continuously, regardless of risk.
In high-risk PV, cytoreductive therapies are recommended up front in addition to aspirin and phlebotomy. The goal is to cytoreduce the patient so that the chances of thrombotic events are decreased, eliminating or decreasing the need for phlebotomies, while also controlling additional counts beyond the red blood cell lineage. Cytoreductive therapy options in the first-line setting include hydroxyurea (HU) or one of the interferon formulations. If there is resistance, refractoriness, or intolerance, NCCN guideline–recommended second-line therapies include ruxolitinib, HU, or one of the interferon formulations not previously used.
For low-risk PV, cytoreductive therapy is occasionally recommended in cases of new thrombotic events, disease-related major bleeding, frequent phlebotomies (generally defined as 3 or more phlebotomies per year), or phlebotomy intolerance. Other reasons for cytoreductive therapy include cases of splenomegaly, progressive thrombocytosis and/or leukocytosis, as well as disease-related symptoms.
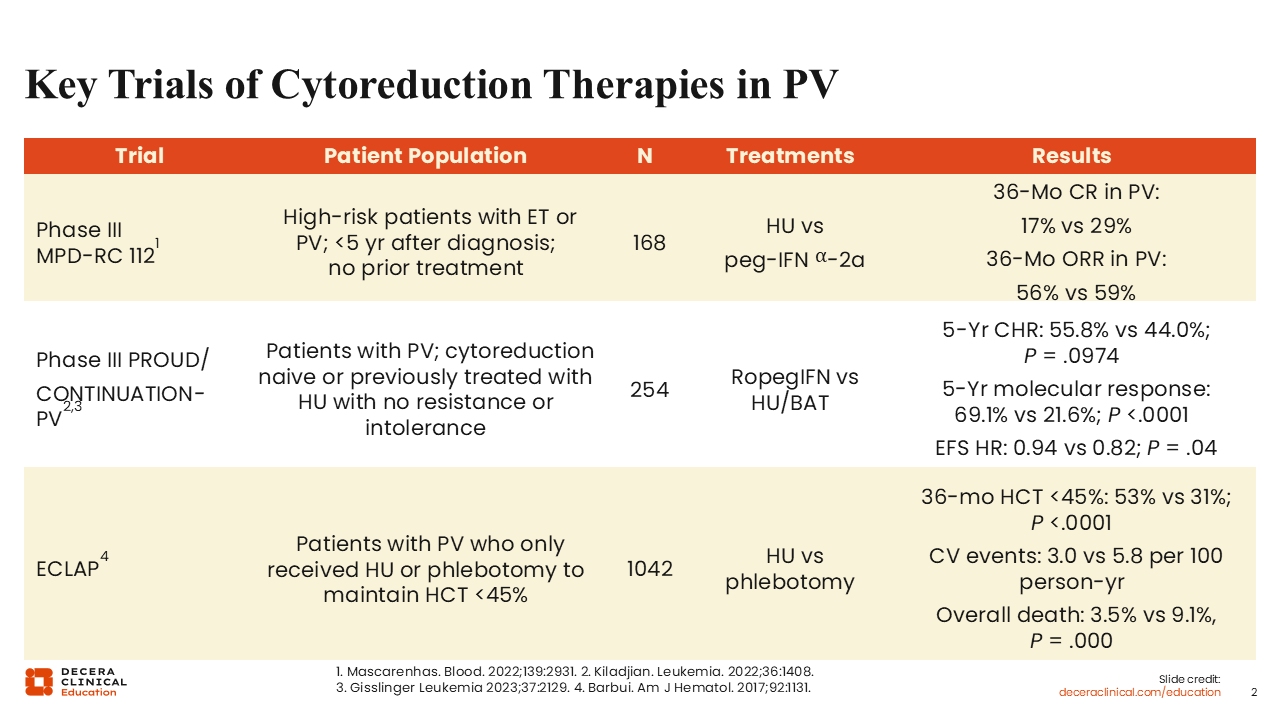
Key Trials of Cytoreduction Therapies in PV
Some key trials of cytoreduction therapies in PV are outlined here. The phase III MPD Research Consortium 112 study evaluated high-risk patients with either PV or ET within 5 years of diagnosis and who had not received prior treatment.4 The treatments studied were HU and peginterferon α-2A. In patients with PV, the complete response (CR) rate and overall response rate were comparable between the 2 treatment groups.
The phase III PROUD/CONTINUATION-PV studies enrolled patients with PV who were cytoreductive therapy naive or previously treated with HU for less than 3 years with no resistance or intolerance.5,6 The studies compared ropeginterferon α-2b vs HU or best available therapy (BAT). Ropeginterferon performed better than HU/BAT, especially after the initial 12 months, as evidenced by the 5-year complete hematologic response rate (55.8% vs 44.0%; P = .0974) and molecular response rate (69.1% vs 21.6%; P <.001), which was not surprising given the reported decrease in variant allele frequency (VAF) of JAK2 V617F over time on ropeginterferon. In addition, event-free survival (EFS) favored the ropeginterferon arm, meaning that ropeginterferon decreased the chances of thrombotic events or transformation.
Finally, the European Collaborative Low-dose Aspirin (ECLAP) study was undertaken to understand the advantage of using aspirin vs not using aspirin in patients with PV.7 In a post hoc propensity score matching analysis, patients who were part of the ECLAP project were compared regarding the ability of HU or phlebotomy to maintain a hematocrit <45%. More than 1000 patients were part of this study. At 36 months, hematocrit control <45% was better achieved with the use of HU compared with phlebotomy. The incidence of fatal/nonfatal cardiovascular events was 3.0 vs 5.8 per 100 person-years, and the rate of overall death was lower in the group that received HU (3.5% vs 9.1%; P = .000).
All of these studies, in addition to the CYTO-PV study,8 which demonstrated that the hematocrit target should be <45% instead of 45% to 50%, form the basis of PV management in practice.
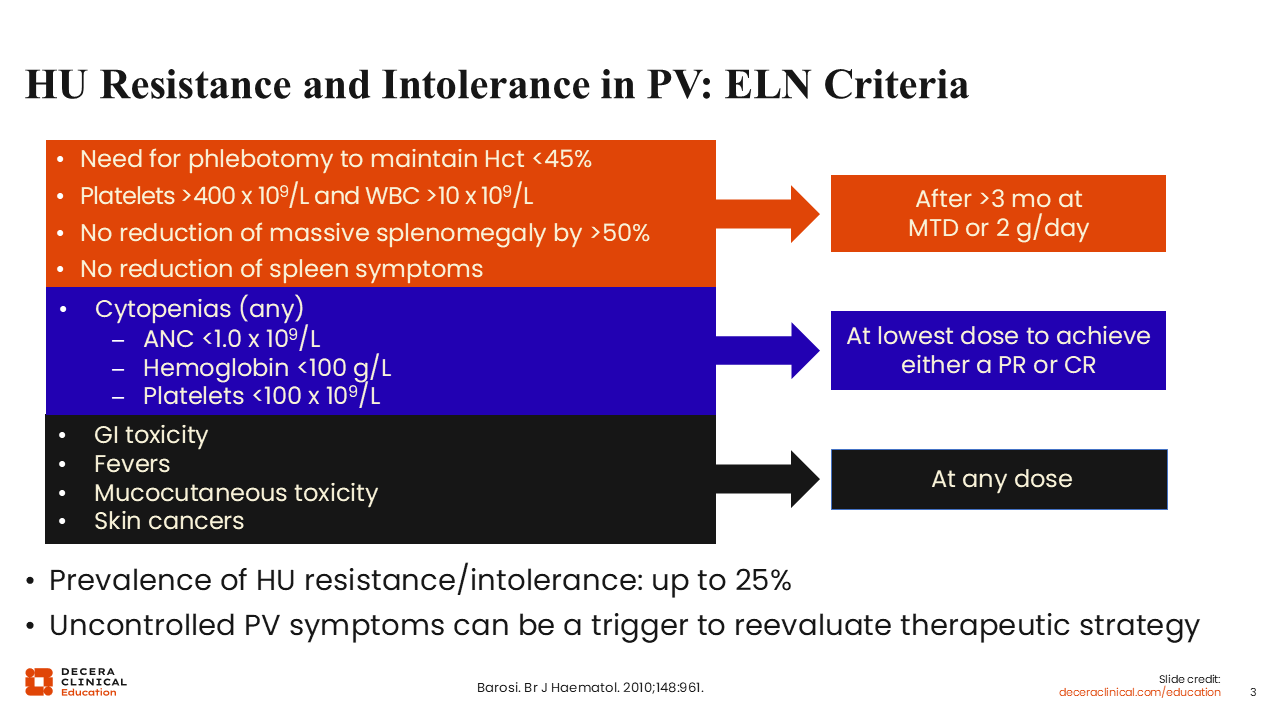
HU Resistance and Intolerance in PV: ELN Criteria
HU resistance and intolerance in PV is currently defined by European LeukemiaNet (ELN) criteria, as shown here.9 These criteria can be subdivided into 3 groups. The first group refers to patients meeting the following criteria: the need for phlebotomy to maintain a hematocrit <45%, elevated platelets and white blood cells, no reduction of massive splenomegaly by at least 50%, or no reduction of spleen symptoms. Overall, these refer more to the refractory nature of or resistance to HU. For these criteria to be met, HU should have been administered at 2 g/day or the maximum tolerated dose for at least 3 months. A patient with cytopenia would also be deemed to have HU intolerance. Finally, the presence of certain adverse events like gastrointestinal toxicity, fevers, mucocutaneous toxicities such as oral ulcers or ankle ulcers, or skin cancers, occurring at any dose of HU, would meet criteria for HU intolerance.
Up to 25% of patients have HU resistance or intolerance, and uncontrolled PV symptoms can be a trigger to reevaluate our therapeutic strategy, meaning that if a patient experiences these symptoms, we should consider switching treatment.
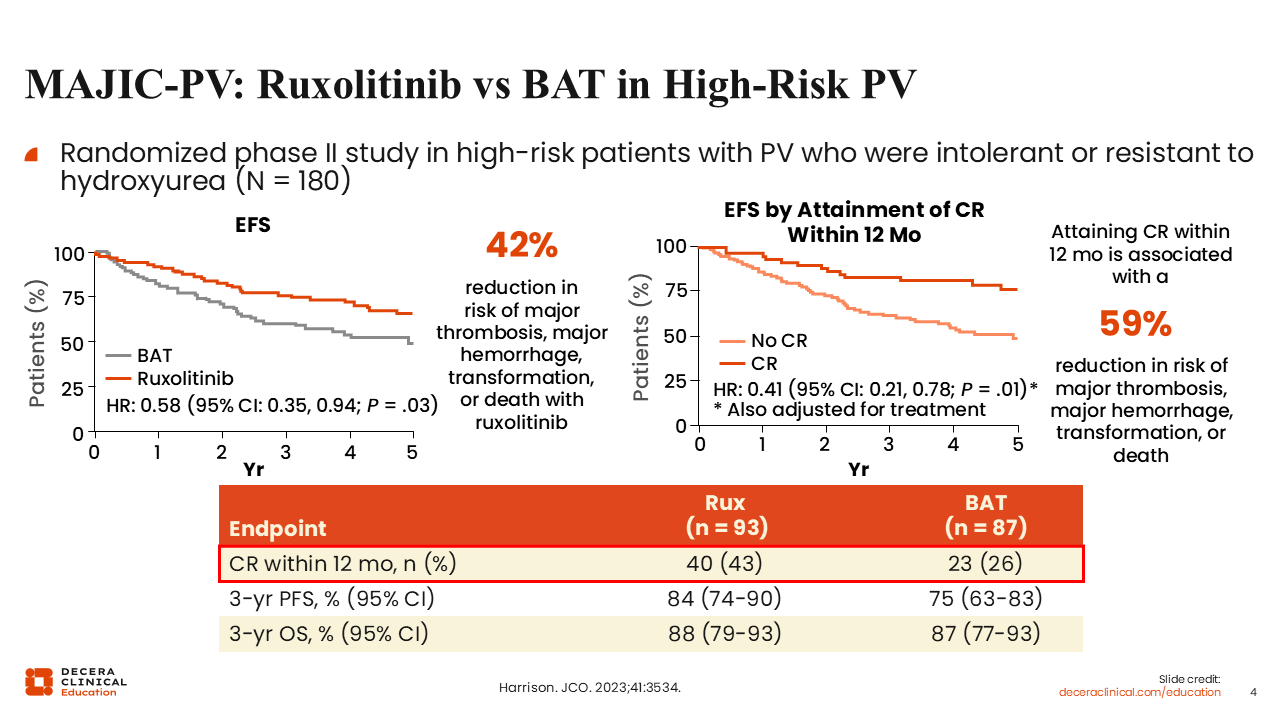
MAJIC-PV: Ruxolitinib vs BAT in High-Risk PV
More recently, the phase II MAJIC-PV study in 180 patients with PV whose disease was resistant or refractory to HU or were intolerant to HU were randomized to ruxolitinib or BAT.10 There was an EFS advantage in favor of ruxolitinib (HR: 0.58; P = .03), where EFS was defined as major thrombotic events, hemorrhagic events, transformation, or death. The data show that switching to a different agent, in this case ruxolitinib, results in better survival than continuing HU. The data also showed that patients who attained a CR within 12 months, regardless of treatment, had better EFS. There were also more molecular responses with the use of ruxolitinib vs BAT. In summary, with ruxolitinib, the CR rates were higher, there were more molecular responses, and EFS was improved by 42% vs BAT including continued HU.
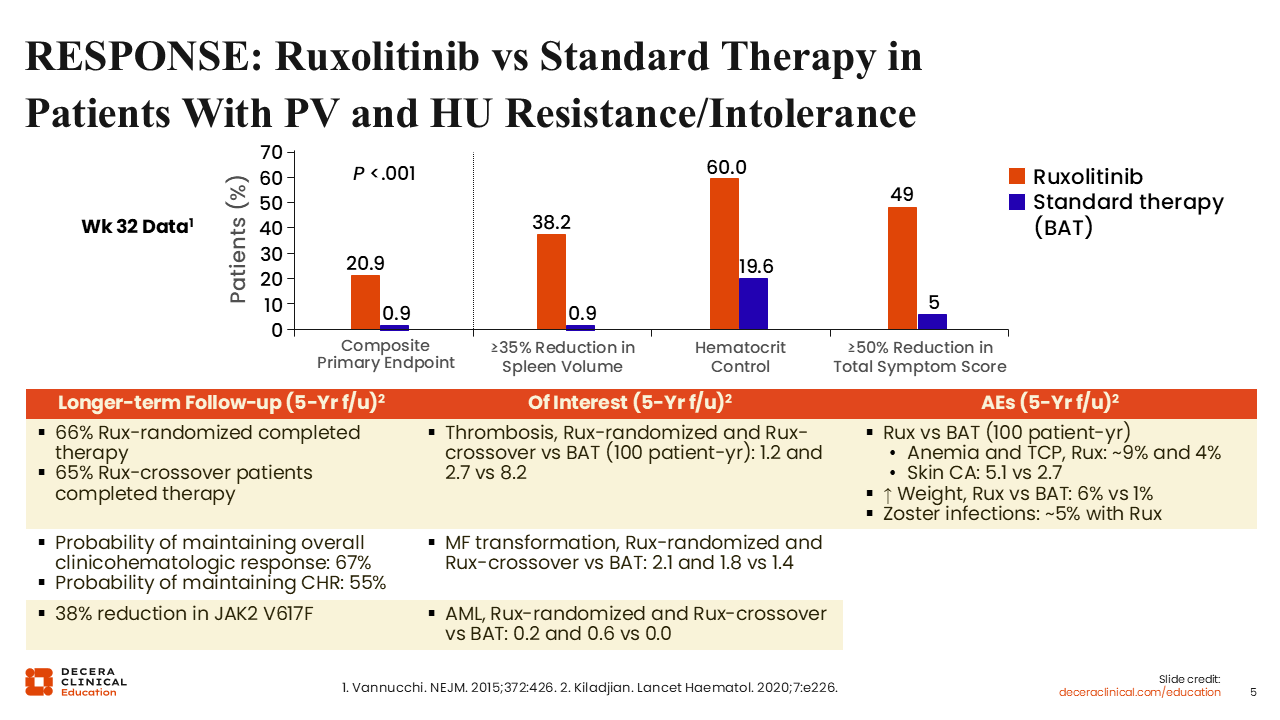
RESPONSE: Ruxolitinib vs Standard Therapy in Patients With PV and HU Resistance/Intolerance
Prior to MAJIC-PV, the results from RESPONSE, a randomized phase III study of ruxolitinib vs standard therapy in patients with PV who had HU resistance or intolerance, were published.11 Unlike MAJIC-PV, in this study, crossover was allowed. The primary endpoint was a composite primary endpoint of a 35% reduction in spleen volume and hematocrit control at 32 weeks, which was met. Response rates were nearly 21% with ruxolitinib vs 1% with standard therapy. When broken down by spleen volume reduction or hematocrit control only, the responses favored ruxolitinib.
Although the study was not designed to look at symptom reduction, it did show that more patients achieved symptom reduction of ≥50% with ruxolitinib (49%) vs standard therapy (5%), which is consistent with what is observed in clinical practice.
We now know that most patients receiving ruxolitinib are able to not only achieve a response but also tolerate the drug over a long period of time.12 Another important finding from this study is the 38% reduction in JAK2 V617F allele burden over time, demonstrating the disease-modifying behavior of ruxolitinib.
In terms of adverse events, reduced rates of thrombotic events favored ruxolitinib (1.2 and 2.7 vs 8.2 thrombotic events per 100 patient-years for patients randomized to ruxolitinib and patients who crossed over to ruxolitinib vs patients randomized to BAT). The incidence of anemia and the risk of skin cancer development were higher with ruxolitinib as were weight increase and herpes zoster reactivation.
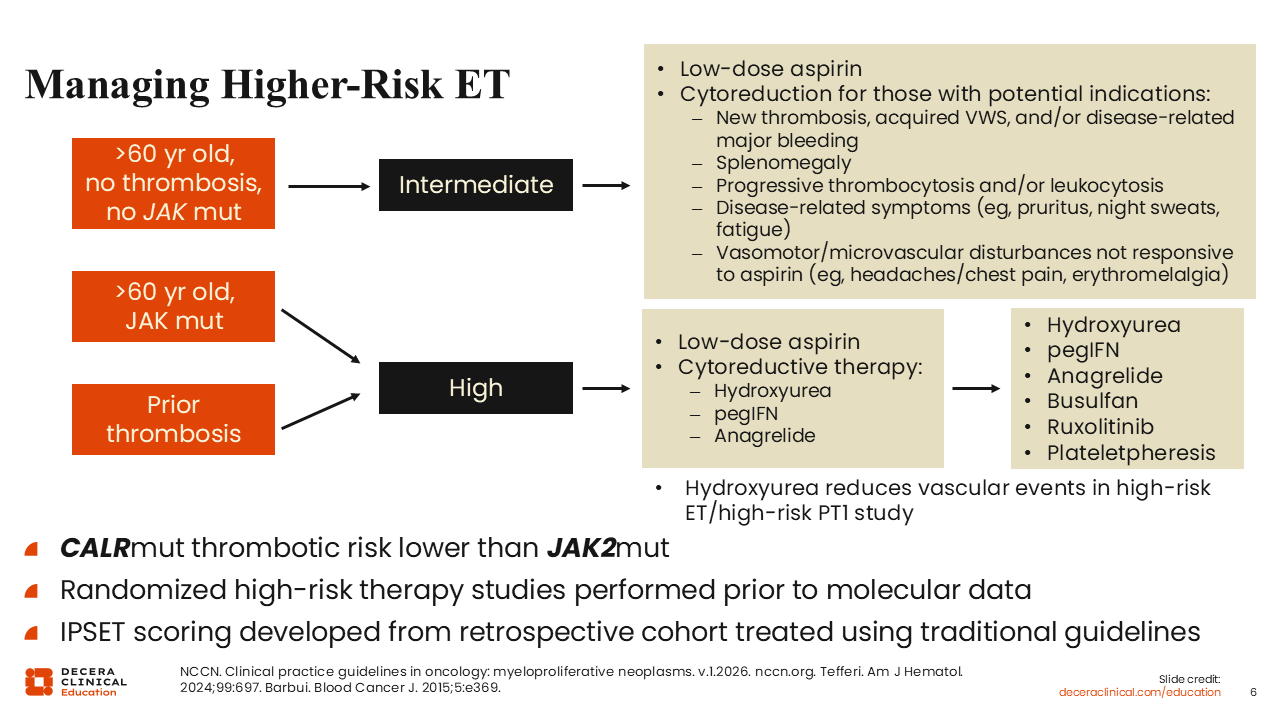
Managing Higher-Risk ET
Let’s shift our focus to ET. This slide illustrates how to manage higher-risk ET.1 Higher-risk ET encompasses the revised IPSET intermediate-risk and high-risk groups.13 Patients who have had a prior thrombotic event or are older than 60 years of age and have a JAK2 mutation are considered high risk. Patients who are older than 60 years but have not had a thrombotic event are at intermediate risk.
For both intermediate-risk and high-risk ET, low-dose aspirin is recommended.1 The difference between these groups is that cytoreductive therapies like HU, peginterferon, or anagrelide are recommended for those at high risk, whereas for patients at intermediate risk, this regimen is recommended only in patients with potential indications like splenomegaly, high symptom burden, new thrombotic events, acquired von Willebrand syndrome, and/or disease-related major bleeding.
Regardless of the treatment setting, in the case of resistance, refractoriness, or intolerance, guidelines recommend switching cytoreductive therapies or plateletpheresis as needed. For most patients, HU, peginterferon, or anagrelide is recommended. Busulfan should be rarely used, as it can be very toxic, and ruxolitinib does not currently have an indication in ET. Plateletpheresis is recommended and reserved for extremely rare cases where platelets are extremely high and an immediate reduction is needed.
It should also be noted, however, that patients with CALR mutations have a lower thrombotic risk than patients with a JAK2 mutation. This was demonstrated in multiple studies14-16 and is the reason why high-risk criteria include JAK2 mutation along with age older than 60 years or a thrombotic event, but not CALR mutation.
These data truly support the use of cytoreductive therapies. Although many key studies in ET were performed prior to the routine use of molecular analysis, newer studies are being done and should allow more personalized treatment.
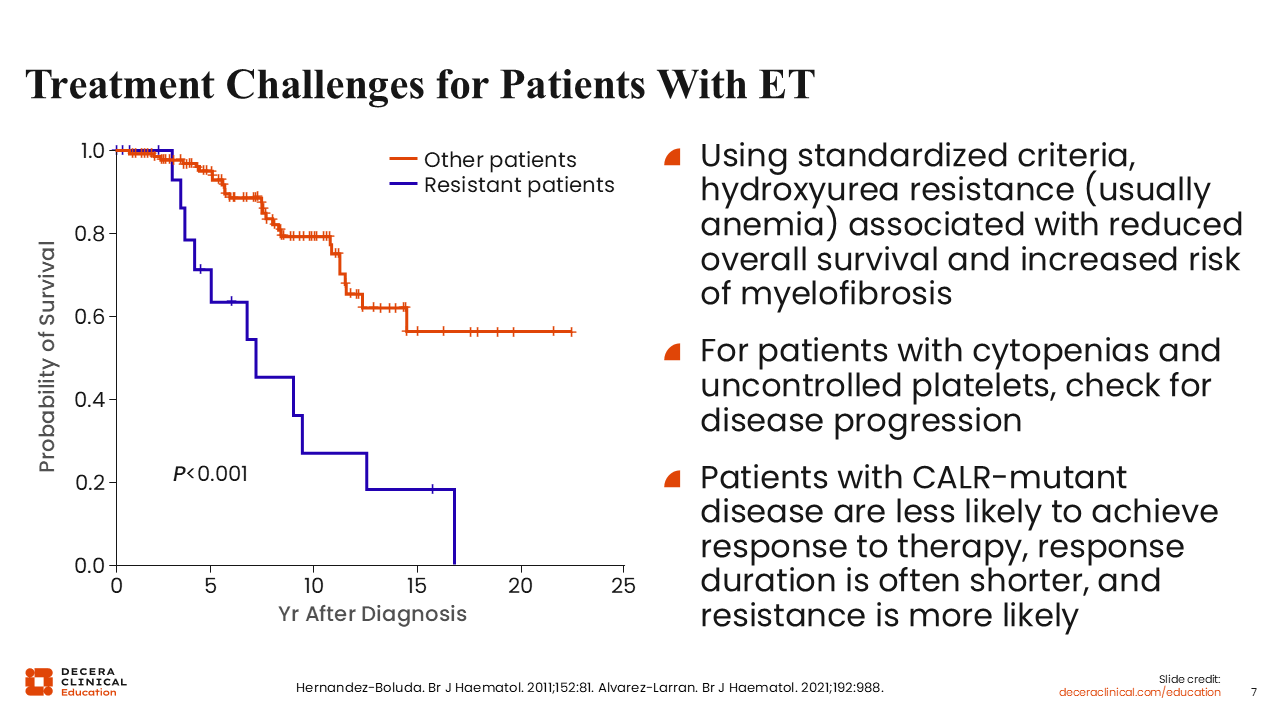
Treatment Challenges for Patients With ET
Much like PV, HU resistance and intolerance can also be challenging in patients with ET. HU resistance not only decreases the chances of survival over time but also is associated with increased risk of progression to myelofibrosis.17 It is important to recognize HU resistance when it occurs so that patients can be switched to a different treatment earlier rather than later.
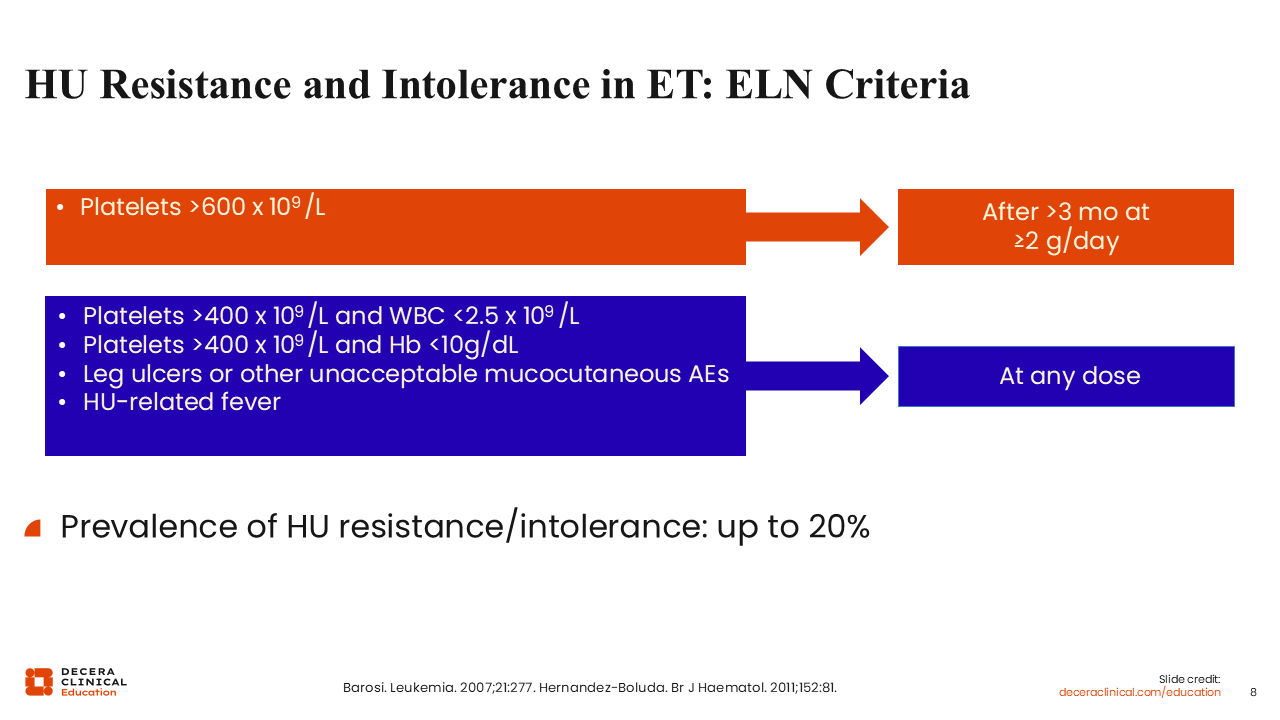
HU Resistance and Intolerance in ET: ELN Criteria
ELN criteria define HU resistance or intolerance as platelets greater than 600 x 109/L after at least 2 g/day of HU or the maximum tolerated dose for a minimum of 3 months.18 Additional criteria include platelets >400 x 109/L and a white blood cell count <2.5 x 109/L, hemoglobin <10 g/dL, the presence of leg ulcers, unacceptable mucocutaneous adverse events, or HU-related fevers at any dose.
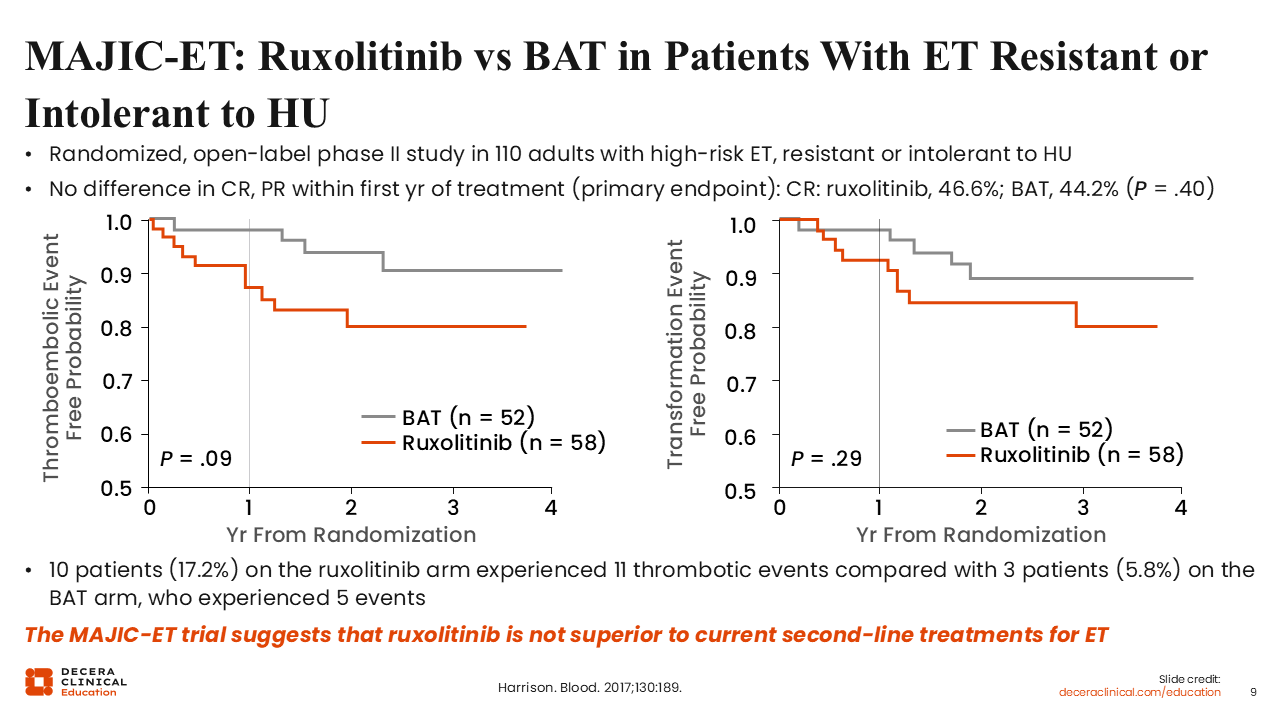
MAJIC-ET: Ruxolitinib vs BAT in Patients With ET Resistant or Intolerant to HU
MAJIC-ET was a sister study to MAJIC-PV.19 In this study, ruxolitinib was compared with BAT in patients with ET who were resistant or intolerant to HU. Although the study results were overall negative, since there was no difference in CR or partial response rates within the first year of treatment, the study did find that ruxolitinib resulted in better thromboembolic event-free probability, although the difference did not reach statistical significance. In addition, there were fewer transformation events, although again the difference did not meet statistical significance, probably because the study was underpowered. Patients who received ruxolitinib had more symptom improvement.
Overall, MAJIC-ET suggested that ruxolitinib was not superior to current second-line treatments for ET, given how the primary endpoint was defined. Nonetheless, as noted, there were some other advantages.
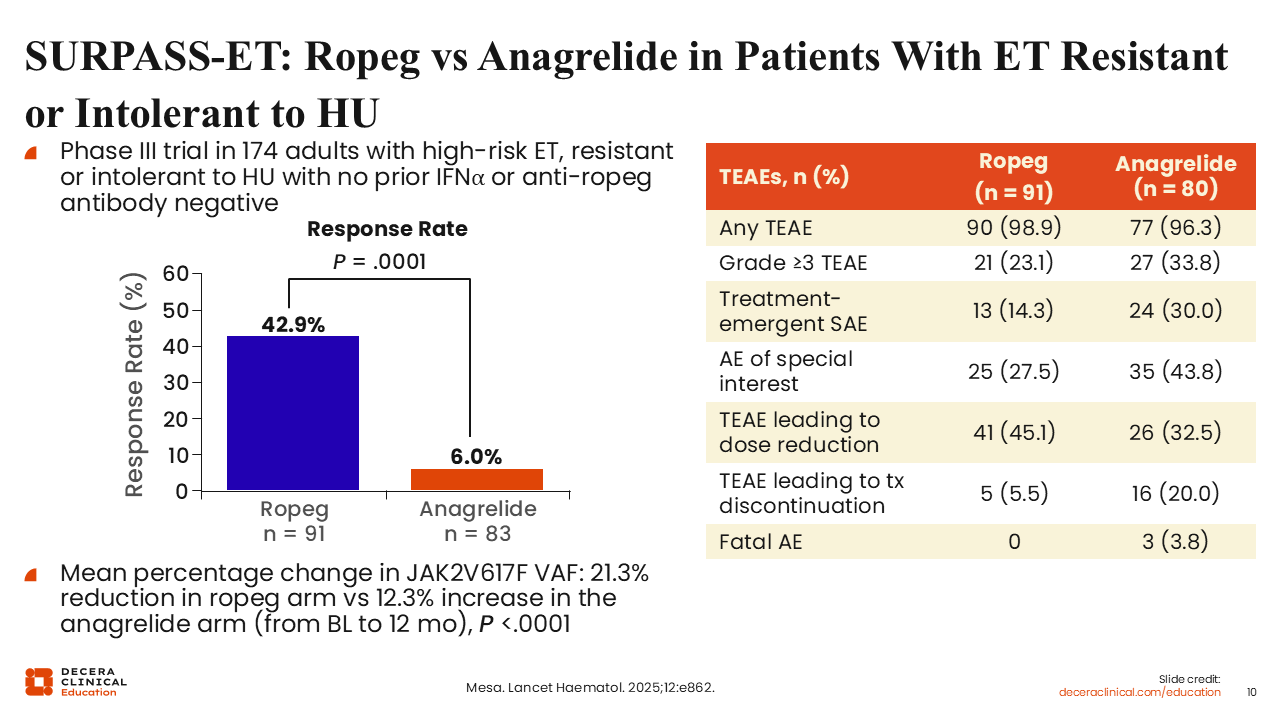
SURPASS-ET: Ropeg vs Anagrelide in Patients With ET Resistant or Intolerant to HU
More recently, SURPASS-ET, a randomized phase III study of ropeginterferon vs anagrelide in patients with ET who were resistant or intolerant to HU, showed that the response rates were higher with ropeginterferon vs anagrelide (42.9% vs 6.0%, respectively).20
Regarding a potential reduction in JAK2 V617F VAF, there was a 21.3% reduction in the ropeginterferon arm vs a 12.3% increase in the anagrelide arm (P <.0001), demonstrating that ropeginterferon is acting at a much deeper level in a disease-modifying fashion, perhaps at the stem cell level, consistent with the known activity of interferon on driver gene-mutant stem cells.
In terms of adverse events, there were more adverse events of special interest with anagrelide. There were some treatment-emergent adverse events that led to dose reductions or dose discontinuations. Dose reductions occurred in slightly more patients on ropeginterferon vs anagrelide. However, it is important to note that the dosing of ropeginterferon in this study differed from the current on-label approach of ropeginterferon used for PV.21
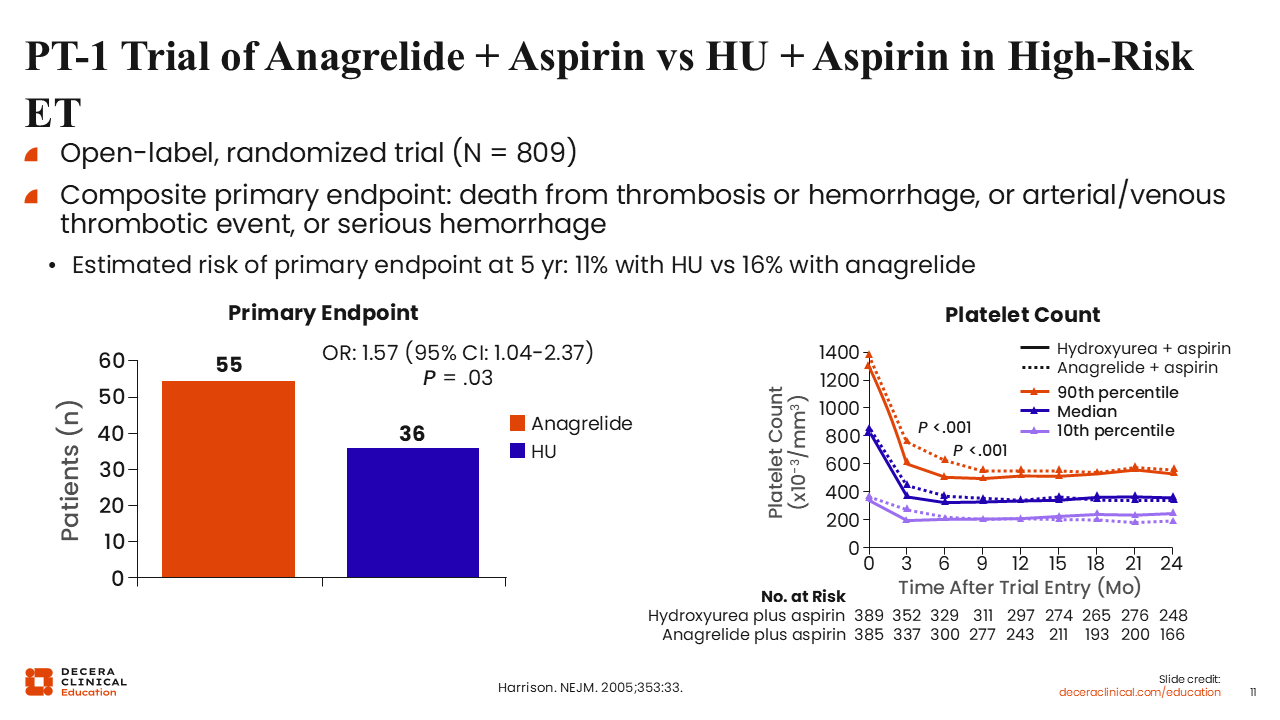
PT-1 Trial of Anagrelide + Aspirin vs HU + Aspirin in High-Risk ET
We also have data from the PT-1 study of anagrelide plus aspirin vs HU plus aspirin in patients with high-risk ET.22 This was a large, randomized trial reported nearly 2 decades ago. The composite primary endpoint was death from thrombosis or hemorrhage, arterial/venous thrombotic events, or any serious hemorrhage. The estimated risk of the primary endpoint at 5 years was 11% with HU vs 16% with anagrelide, demonstrating that both drugs were active. More patients treated with anagrelide met the primary endpoint. Overall, this demonstrated that HU plus low-dose aspirin is superior to anagrelide plus low-dose aspirin for patients with ET at high risk for vascular events.
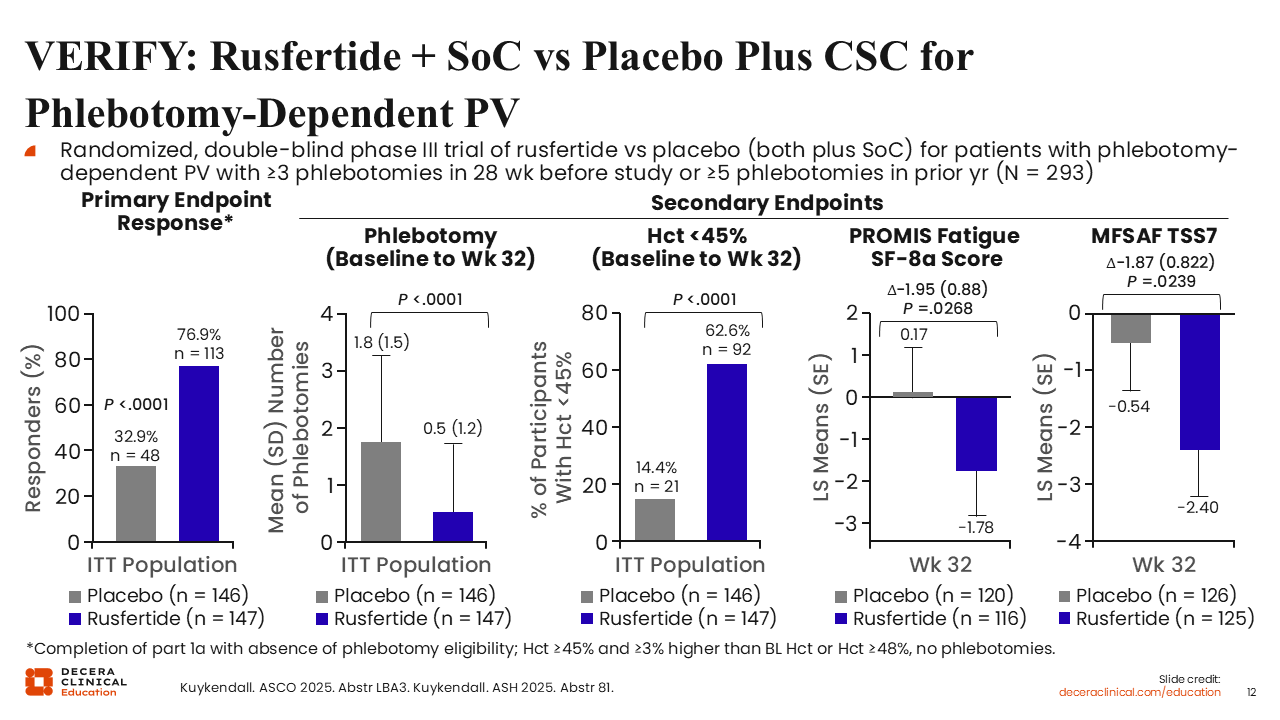
VERIFY: Rusfertide + SoC vs Placebo Plus CSC for Phlebotomy-Dependent PV
Now I want to discuss emerging therapies, beginning with those for PV. Rusfertide is a hepcidin mimetic that reduces iron availability to the bone marrow, thus affecting erythropoiesis. Rusfertide was examined in the phase II REVIVE trial23 and later in the phase III VERIFY study in patients with phlebotomy-dependent PV.24,25 Participants were randomized in a double-blinded fashion to the addition of rusfertide or placebo to standard of care, which was phlebotomies and aspirin or cytoreductive therapies. In fact, nearly 55% of patients in each arm were receiving concurrent cytoreductive therapy. The primary endpoint, proportion of participants achieving a clinical response, was met in more patients receiving rusfertide vs placebo (76.9% vs 32.9%, respectively; P <.0001).24
Equally interesting and perhaps of more importance, all the key secondary endpoints were also in favor of rusfertide. For example, phlebotomy requirement was significantly decreased in patients on rusfertide vs standard of care (mean phlebotomies: 0.5 vs 1.8; P <.0001). Significantly more patients receiving rusfertide maintained a hematocrit <45% (62.6% vs 14.4%; P <.0001), and patient-reported outcome data also favored the use of rusfertide.25 Rusfertide is currently undergoing regulatory review by the FDA.
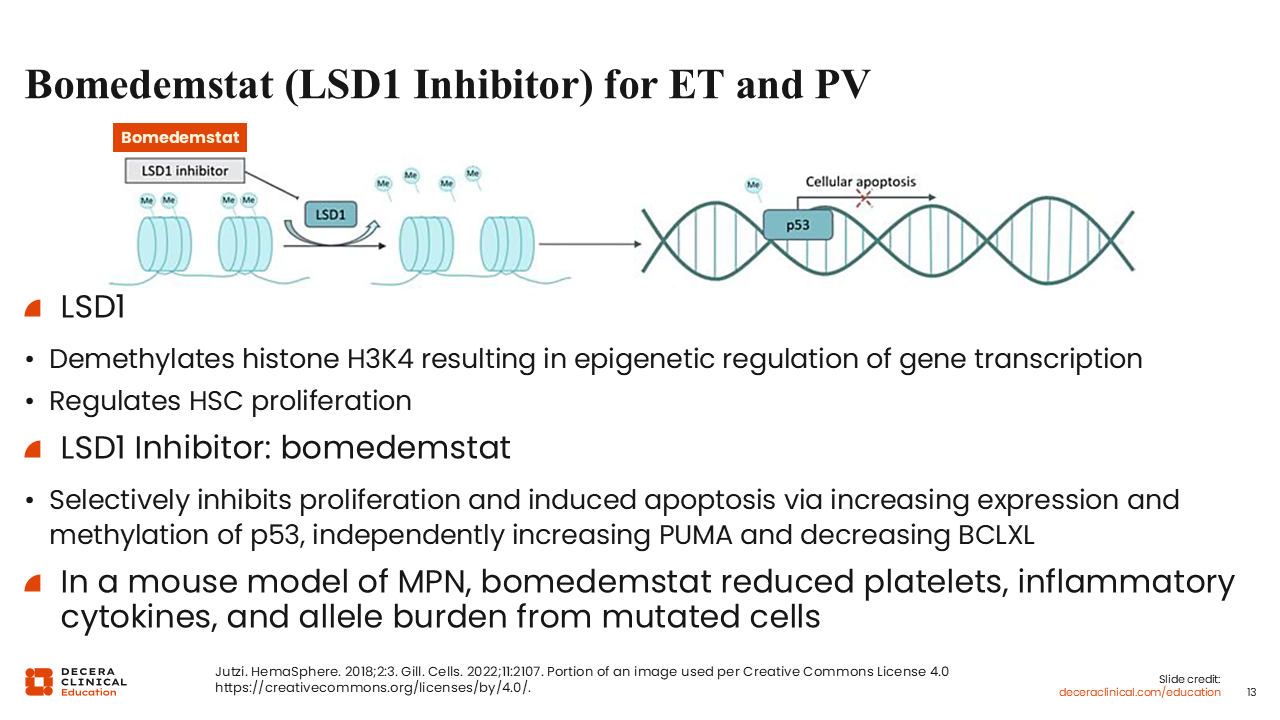
Bomedemstat (LSD1 Inhibitor) for ET and PV
Another emerging agent is bomedemstat, an LSD1 inhibitor being developed for both ET and PV. LSD1 levels are increased in patients with myeloproliferative neoplasms (MPNs).26 LSD1 demethylates histone H3K4, which leads to changes in epigenetic regulation of gene transcription.27,28 In many ways it regulates hematopoietic stem cell proliferation. When LSD1 is inhibited, the BCL2 family of proteins is altered. In particular, PUMA levels are increased and BCLXL levels are decreased, shifting the balance toward apoptosis. Herein lies the rationale behind using an LSD1 inhibitor: LSD1 levels are high in MPN samples and inhibiting LSD1 leads to apoptosis. In mouse models of MPNs, bomedemstat reduced platelets, inflammatory cytokines, and the allele burden from mutated cells.
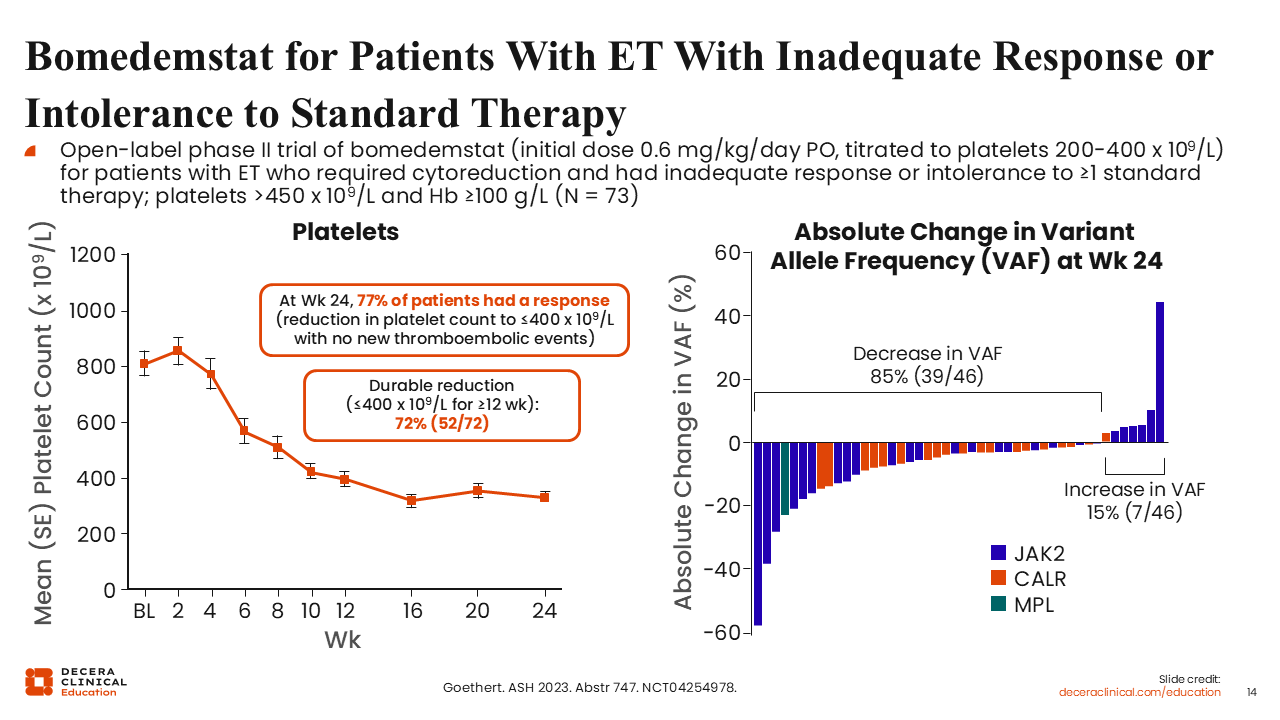
Bomedemstat for Patients With ET With Inadequate Response or Intolerance to Standard Therapy
In this open-label phase II trial (NCT04264978), patients who had an inadequate response or were intolerant to their standard therapy were given bomedemstat at an initial dose of 0.6 mg/kg/day orally.29 It is important to keep in mind that bomedemstat was titrated to keep platelets in the 200 to 400 x 109/L range. Platelets decreased in a remarkable fashion. For example, at Week 24, 77% of patients had a response, meaning a platelet count ≤400 x 109/L. This finding is remarkable, and the responses were durable.
Of equal importance, there was a reduction in the VAF of driver gene mutations in the majority (85%) of patients.
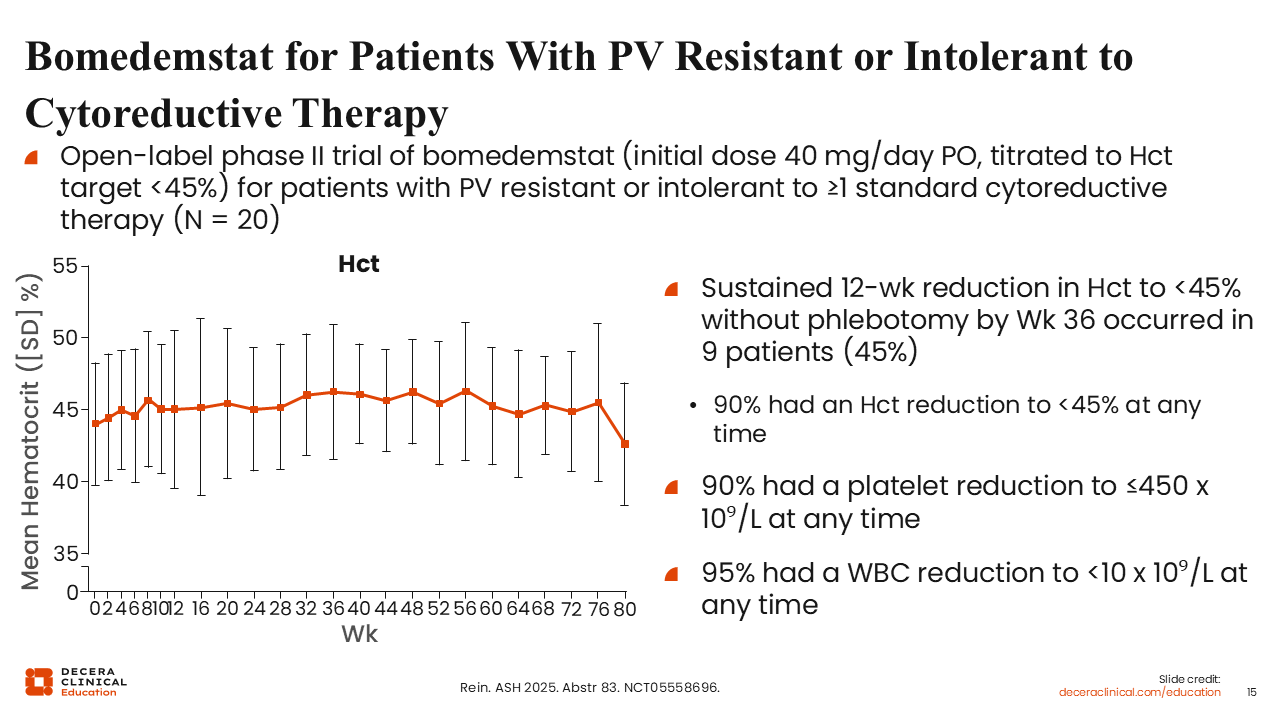
Bomedemstat for Patients With PV Resistant or Intolerant to Cytoreductive Therapy
Another open-label phase II trial (NCT05558696) of bomedemstat in patients with PV resistant or intolerant to at least 1 cytoreductive therapy focused on hematocrit control.30 Bomedemstat was initiated at 40 mg/day orally and titrated to keep the hematocrit <45%. The sustained 12-week reduction in hematocrit to <45% was achieved in 45% of patients. There was also a reduction in white blood cell count, which is important since multiple studies support that an increased white blood cell count leads to a higher thrombotic risk, even when the hematocrit is controlled.31,32 In summary, bomedemstat helped to decrease platelet and white blood cell counts in addition to decreasing the hematocrit for most patients.
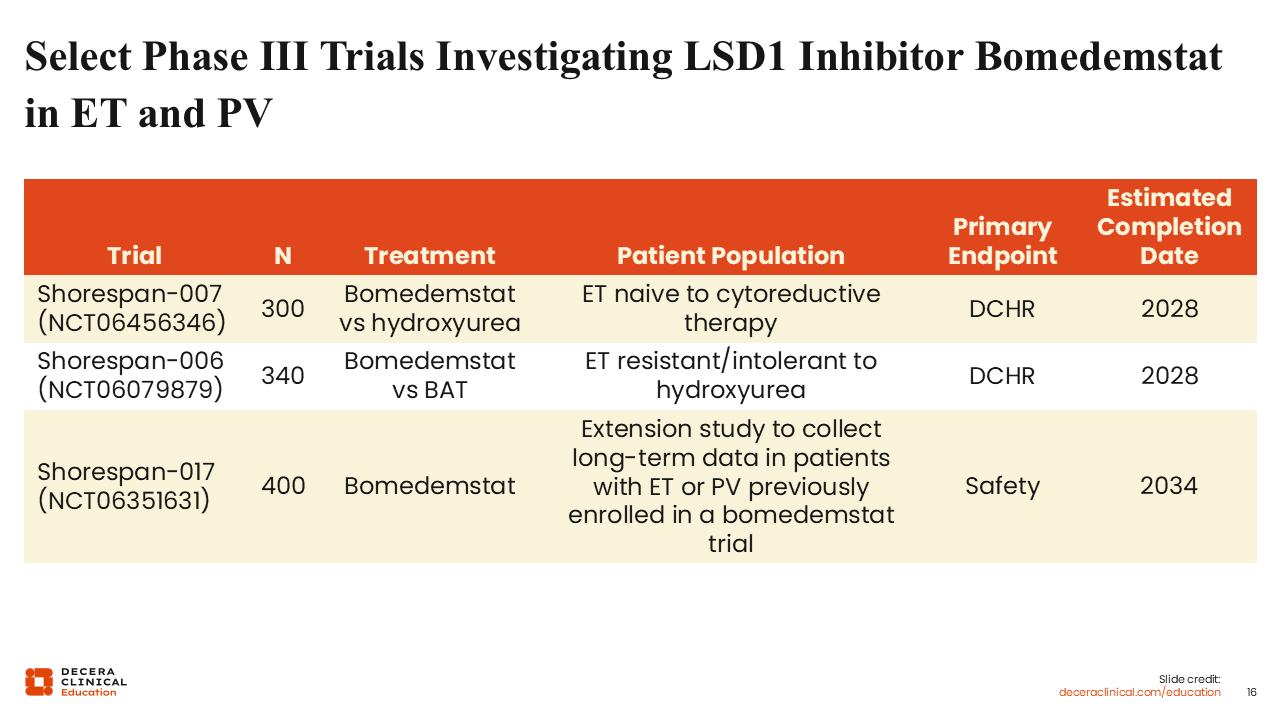
Select Phase III Trials Investigating LSD1 Inhibitor Bomedemstat in ET and PV
Bomedemstat is currently being studied in a few phase III trials, including Shorespan-007 and -006, which are enrolling patients with ET who are either treatment naive to cytoreductive therapy or are resistant or intolerant to HU, respectively. The primary endpoint is durable complete hematologic response. There is also the Shorespan-017 study, an extension study to collect long-term data in patients with ET or PV who were previously enrolled in a bomedemstat trial—basically a rollover trial with a primary endpoint of safety.
MANIFEST (Arm 4): Phase II Trial of BET Inhibitor Pelabresib in High-Risk ET
Another emerging agent is pelabresib, a small molecule BET inhibitor. This slide provides an overview of MANIFEST, a phase II study of pelabresib, specifically arm 4.33 In this arm, 20 patients with symptomatic, high-risk ET resistant or intolerant to HU and with platelets >600 x 109/L were enrolled. This study found that pelabresib helped reduce platelet counts and resulted in symptom improvement. Some of the adverse events seen (eg, diarrhea and dysgeusia) were in line with those observed in patients with myelofibrosis treated with pelabresib.

CALR Targeting Agents Being Investigated in ET
Other exciting drugs also being studied include CALR-targeting antibodies, which are being investigated in the high-risk ET setting. Preliminary data for INCA33989, a monoclonal antibody, found that the maximum tolerated dose was not reached, and the majority of patients (83.3%) receiving a dose of 400-2400 mg achieved a complete hematologic response. Also of note were the data regarding the reduction of 25% or more in the mutant CALR VAF that was observed in 52% of patients.34
There is also the bispecific antibody, JNJ-885499682, which targets mutant CALR and CD3, that is currently being investigated in patients with MPNs.35

Selective JAK Inhibitors in ET
Finally, the selective JAK2 V617F–specific inhibitor INCB160058 is currently being studied in ongoing phase I trials in patients with MPNs harboring JAK2 V617F.36 INCB160058 is a high-affinity JH2-binding inhibitor. The premise here is that mutant clones would be targeted specifically while preserving normal hematopoiesis. With this selective targeting, higher doses of the drug could be possible, and VAF could be controlled by targeting the mutant clones while not negatively affecting safety.
It is an exciting time as the treatment landscape for PV and ET are continuing to evolve, hopefully providing more effective therapeutic options for our patients.
