CE / CME
Clinical Focus in Paroxysmal Nocturnal Hemoglobinuria: New Strategies for Evading the Complement Cascade
ABIM MOC: maximum of 0.75 Medical Knowledge MOC point
Physicians: Maximum of 0.75 AMA PRA Category 1 Credit™
Released: March 10, 2026
Expiration: September 09, 2026


Activity
The Role of Complement Activation in PNH
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
PNH is a rare blood disorder characterized by hemolysis of RBCs, resulting in anemia and thromboembolic events. To understand PNH, we need to first understand the complement cascade. This figure shows a schematic representation of how complement is activated through the classical pathway, the lectin pathway, and the alternative pathway.1 In PNH, the more important component is the alternative pathway that is continuously active through a spontaneous, or “tickover” mechanism. Here, C3 undergoes hydrolysis and forms C3 convertase that includes Factor B and Factor D, resulting in amplification of C3 activation. C3 convertase then works on C5 and activates C5 convertase, which ultimately results in the formation of a membrane attack complex that causes RBC destruction.
Normal cells have mechanisms to protect themselves against complement activation. These include CD55, which can block C3 and C3a, as well as CD59, which is able to block the terminal pathway. These 2 proteins are anchored to the cell membrane through a GPI anchor, and this is where the pathophysiology of PNH becomes much more interesting.

PNH Molecular Pathogenesis: PIGA Mutation and GPI Anchors
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
For PNH to develop, multiple events must occur simultaneously. The first step is acquisition of ≥1 mutations in the PIGA gene, which is a component in the first step of GPI biosynthesis. Mutations that either block production of PIGA or reduce its expression can result in a partial or complete deficiency of PIGA expression in cells. Consequently, there is reduced or absent expression of the GPI-anchored proteins CD55 and CD59. The PIGA mutation occurs in hematopoietic stem cells because the phenotype is seen in all types of hematopoietic cells. When the mutant stem cell expands, it contributes substantially to hematopoiesis. Although the mechanism is elusive, the expansion of mutant stem cells is likely triggered by extrinsic immune-mediated selection for this population.2,3
In PNH, complement is continuously active at a very low level and activated C3 is membrane bound. CD55 and CD59 are lost, and RBCs do not have any other mechanism to control complement activity. RBCs lack the complement regulating CD46 and CR1 membrane proteins, which appear to function differently on RBCs compared with nucleated cells.
Clinical Pathogenesis of PNH
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
Hemolysis is continuous in PNH and a major contributor of its pathogenesis. In untreated patients, intravascular hemolysis mainly occurs, whereas patients treated with a C5 inhibitor can develop extravascular hemolysis. The resulting damage to organs is multifactorial.4 For example, in the renal system, free hemoglobin is filtered by the kidneys and incurs a loss of nitric oxide and microthrombi. Nitric oxide depletion is critical for other symptoms related to PNH. The presence of free hemoglobin depletes nitric oxide, and that can result in smooth muscle spasms, abdominal pain, esophageal spasm, and erectile dysfunction. Nitric oxide is also critical for endothelial function, and therefore, the loss of nitric oxide can result in increased risk of thrombosis. With PNH, thrombosis is also multifactorial because of platelet dysfunction and activation of the procoagulant system.5 Pulmonary hypertension is a partial consequence of loss of nitric oxide from the endothelium.

Disease Presentation: Suspicion of PNH
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
PNH presents with a variety of symptoms because of hemolytic anemia. Therefore, patients may be short of breath, fatigued, or jaundiced. Having dark urine is a major characteristic that gives the disease its name.6,7
If PNH is suspected, laboratory tests should be undertaken to confirm the presence of anemia, hemolysis with an increased reticulocyte count, increased bilirubin, and elevated LDH, which is typical of intravascular hemolysis. The patient will often have low haptoglobin and may have urine hemosiderin. The Coombs test in untreated PNH is negative. It is important to distinguish PNH from the more common warm autoimmune hemolytic anemia, in which the Coombs test is positive. I also obtain a D-dimer to evaluate the risk of thrombosis. Any patient with these signs and symptoms should be tested for PNH. I also screen for PNH in any patient who has a bone marrow failure syndrome. Patients who have unusual and otherwise unexplained symptoms, including muscle spasms, unexplained renal insufficiency, abdominal pain in the context of hemolytic anemia, erectile dysfunction, pulmonary hypertension, or unexplained iron deficiency, again in the context of ongoing hemolysis, should trigger testing for PNH.

Diagnosis of PNH: Flow Cytometry
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
Diagnosis of PNH with the use of flow cytometry is straightforward. An example of a flow cytometric analysis of a patient with PNH is shown here. The process happening is clonal and affecting RBCs, monocytes, and granulocytes.
The slide shows a patient with 2 populations of PNH cells. At the top is a histogram showing a peak with a large number of normal RBCs with high-level expression of GPI-anchored proteins, like CD59. At the top right is a scatter plot that shows the expression and ratio of RBCs with CD59, the GPI-anchored protein lost in PNH, and CD235a, which is glycophorin A expressed on the surface of RBCs. The scatter plot shows RBCs with an intermediate population of some expression of GPI-anchored proteins as well as cells without any GPI expression.
The way we test for PNH using flow cytometry is standardized, and there are various ways to look for PNH phenotype. This includes the use of the FLAER reagent that directly interacts with GPI anchor, but we also look for GPI-anchored proteins such as CD14, CD24, and so on. For example, in the lower middle panel, there are 3 populations of cells. We have cells that bind to FLAER, indicating that GPI is expressed, shown as the green dots. Then there is a large population of cells, shown as turquoise dots, that do not express either CD24 or interact with the FLAER reagent. These are the cells that do not express GPI anchor, or CD24, which is another GPI-anchored protein. Then there is a small population of cells in magenta that have partial deficiency. The same phenotype is seen with monocytes, in the far right lower panel. Then we have the same phenotype with RBCs in the lower left panel.
Typically, clone size is measured based on the estimates of monocytes and granulocytes, which should be similar. In the untreated patient, the clone size based on RBCs is typically much lower because the red cells are being destroyed.

PNH: Differential Diagnosis and Recommended Testing
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
The differential diagnosis for PNH includes other causes of hemolytic anemia.7 Typically, PNH is an acquired problem, so the patient will not have a history that is compatible with hereditary hemolytic anemia, such as long-standing hemolysis or a family history of the disease. Autoimmune hemolytic anemia can often be excluded with a Coombs test. Thalassemia syndrome or traumatic hemolysis should also be excluded, with the latter usually being obvious. Other causes of anemia or bone marrow failure syndromes should also be assessed.
The following tests are relevant for establishing the diagnosis: complete blood count, reticulocyte count, metabolic panel, LDH, haptoglobin, Coombs test, D-dimer and flow cytometry.

Therapy for Classic Hemolytic PNH
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
Once the diagnosis of PNH has been made, the next question is whether the patient requires therapy; not every patient with PNH needs to be treated. The following factors should be taken into consideration7: clone size, hemoglobin level, transfusion history, and PNH phenotype (type II vs type III). For example, patients with type II PNH cells tend to have milder hemolysis and may or may not require therapy.
Decisions on the need for therapy are based on the patient’s prior thrombotic history, the severity of the anemia, and the presence of comorbidities such as kidney dysfunction or pulmonary hypertension.
Treatment of PNH requires complement-directed therapy. If the patient has no history of thrombosis, then anticoagulation is generally not required with modern complement-directed therapy. It is important to provide appropriate supportive care, particularly immunizations. Antibiotic prophylaxis and, in the appropriate setting, iron and vitamin supplementation may also be given.
Patient education is fundamental to the management of PNH so that patients understand the disease, the rationale behind therapy and associated risks, and when to alert the care team if such problems arise.

PNH: Current Classification and Standard of Care
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
PNH is categorized as classic hemolytic PNH, PNH with bone marrow failure syndrome, or subclinical PNH.7 Typically, patients with classic hemolytic PNH have a larger, GPI anchor–deficient granulocyte clone size, defined as >20% or >50%. If the clone is >20%, patients are likely to require therapy. PNH with bone marrow failure syndrome tends to feature small clones, generally 20% to 30%, but often smaller. The presence of a PNH clone in a patient with a bone marrow failure syndrome, such as aplastic anemia, usually means that the bone marrow failure is acquired rather than inherited. Patients with subclinical PNH have a small PNH clone <10% in size and usually do not require therapy.
Current therapy for PNH can be divided into the C5 inhibitors eculizumab; the first drug approved for PNH, ravulizumab; and more recently, crovalimab. These agents are also known as terminal complement inhibitors. Then the group known as proximal complement inhibitors includes pegcetacoplan that targets C3 and the Factor B inhibitor iptacopan, which are approved as single agents, whereas danicopan, a Factor D inhibitor, must be given in combination with a C5 inhibitor.

Eculizumab and Ravulizumab in PNH
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
Eculizumab was the first drug that was approved for use in PNH.8 The phase III SHEPHERD trial of eculizumab in patients with PNH showed that with initiation of therapy, the RBC clone size increases. This occurs because the RBCs are protected from complement-mediated hemolysis, which improves hemoglobin, and the Functional Assessment of Chronic Illness Therapy (FACIT)-Fatigue score improves.9
The approval of ravulizumab for PNH was based on findings from the 301 and 302 phase III trials of eculizumab vs ravulizumab in C5 inhibitor–naive and C5 inhibitor–-experienced patients, respectively.10,11 Ravulizumab was shown to be noninferior to eculizumab since a larger percentage of patients receiving ravulizumab reached normalization of LDH during the 26-week treatment period of the trial.10 Ravulizumab is a more convenient agent than eculizumab because it is given every 8 weeks rather than every 2 weeks.

Recent Practice-Changing Approvals of Complement Inhibitors in PNH
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
Several new agents have been approved for PNH in the last few years. These are the proximal inhibitors, pegcetacoplan, which targets C3; iptacopan, which targets Factor B; and danicopan, which targets Factor D. Factor D is a cofactor with Factor B in the alternative pathway as the C3 convertase amplification loop. Iptacopan and danicopan are oral agents, whereas pegcetacoplan is given subcutaneously.1

PEGASUS: Pegcetacoplan vs Eculizumab in Patients With PNH
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
Despite the availability of C5 inhibitors, a significant percentage of patients with PNH, perhaps 25% to 30%, continue to be anemic with a high reticulocyte count, often requiring transfusions. Extravascular hemolysis occurs when the accumulation of C3 on the surface of RBCs renders them opsonized for destruction in the reticuloendothelial system of the liver and spleen. Pegcetacoplan was developed with the aim of mitigating both intravascular and extravascular hemolysis.
Pegcetacoplan is a cyclic peptide given subcutaneously twice weekly as a self-administered therapy. PEGASUS, an international, open-label, randomized phase III trial, compared pegcetacoplan with eculizumab in patients with preexisting PNH who had a persistent anemia on stable doses of eculizumab for at least 3 months before screening.12 All participants received a 4-week run-in with pegcetacoplan 1080 mg twice weekly plus eculizumab before randomization to 16 weeks of pegcetacoplan or continued eculizumab. After this randomized period, all patients were eligible for 32-week open-label pegcetacoplan.
The primary endpoint was mean change in hemoglobin level from baseline to Week 16. Secondary endpoints included the need for transfusion and the impact on LDH level, FACIT-Fatigue score, and other safety markers.

PEGASUS: Mean Hemoglobin Levels of Pegcetacoplan vs Eculizumab
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
Mean hemoglobin was 8.7 g/dL at baseline.12 At Day 1, following the 4-week run-in period in which pegcetacoplan was added to eculizumab, mean hemoglobin improved to 11.9 g/dL. At the end of the 16-week randomized treatment period, patients receiving pegcetacoplan maintained their hemoglobin around this level, whereas, as expected, the patients who were randomized back to the eculizumab returned to their baseline level. At Week 16, adjusted least-squares mean change in hemoglobin vs baseline was 2.37 g/dL with pegcetacoplan and -1.47 g/dL with eculizumab. The mean difference between treatment arms was 3.84 g/dL (95% CI: 2.33-5.34; P <.001), indicating superiority of pegcetacoplan vs eculizumab.
The need for transfusion was greater among patients receiving eculizumab. Therefore, the study confirmed that proximal inhibition can result in an improvement in hemoglobin and illustrated the importance of extravascular hemolysis as a mechanism for ongoing and persistent anemia that may require transfusions.

Long-term Efficacy of Pegcetacoplan in Patients With PNH
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
The PRINCE trial was a study of pegcetacoplan in treatment-naive patients with PNH. Combined results illustrate the long-term efficacy of pegcetacoplan in patients. The plotted data show the extended results from the PEGASUS trial as well as the extended data from the PRINCE trial. Patients treated with pegcetacoplan had an increase in their rate of transfusion avoidance. After almost 3 years, patients continued to benefit from pegcetacoplan therapy with stabilization of their hemoglobin at a substantially high levels, often close to normal levels, which indicates that patients generally do not lose the response to therapy.13

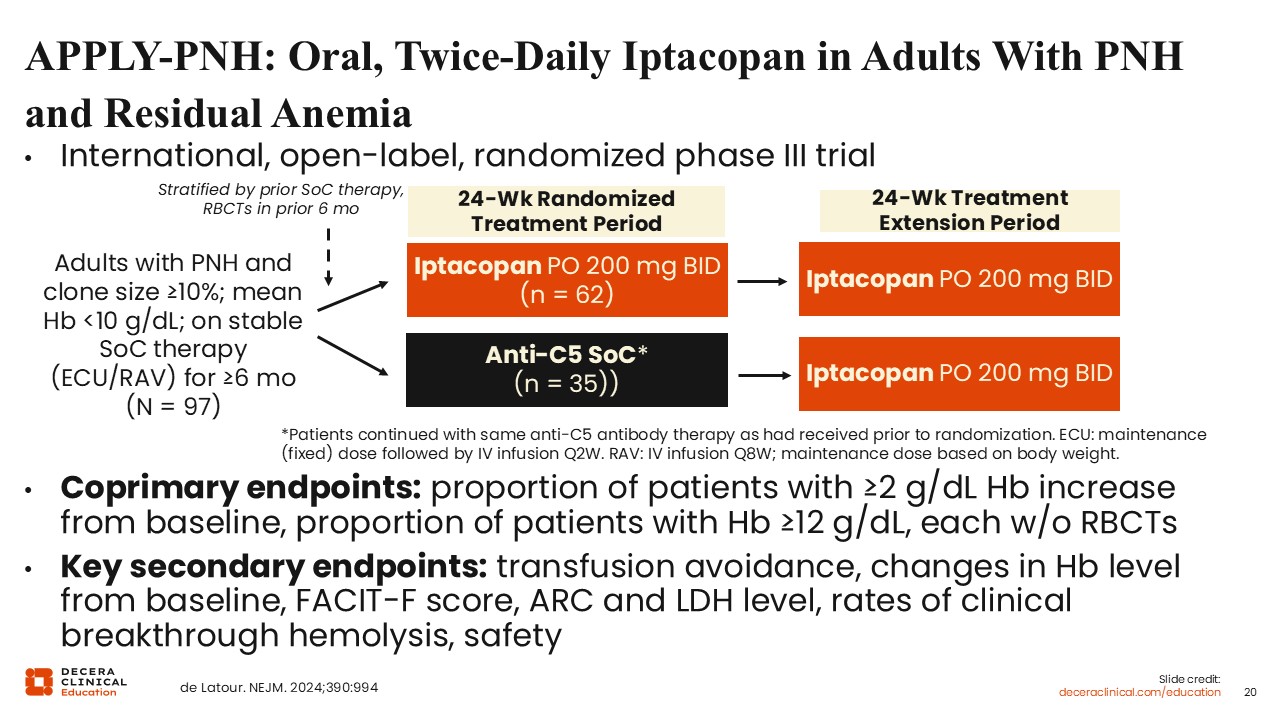
APPLY-PNH: Oral, Twice-Daily Iptacopan in Adults With PNH and Residual Anemia
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
Iptacopan, an oral proximal complement inhibitor targeting Factor B in the alternative pathway, has been studied in patients with PNH in a number of trials. APPLY-PNH was an international, open-label, randomized phase III trial of oral, twice-daily iptacopan in adults with PNH and residual anemia.14 Patients with PNH who had been receiving a C5 inhibitor (eculizumab or ravulizumab) for at least 6 months, with clone size ≥10% and mean hemoglobin <10 g/dL were randomized to continued C5 inhibitor or iptacopan 200 milligrams twice daily for 24 weeks. After the 24-week randomization period, the patients in the C5 standard-of-care arm could switch to iptacopan in a 24-week extension period.
Coprimary endpoints were the proportion of patients with ≥2 g/dL hemoglobin increase from baseline, and the proportion of patients with hemoglobin ≥12 g/dL, each without the need for RBC transfusions. Key secondary endpoints were transfusion avoidance, changes in hemoglobin level from baseline, FACIT-F score, absolute reticulocyte count (ARC), LDH level, rates of clinical breakthrough hemolysis, and safety.15
APPLY-PNH: Results of Iptacopan on Anemia and Mean Hb
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
After the 24-week randomized treatment period, treatment with iptacopan was associated with a substantial improvement in hemoglobin levels. Forty-two of 60 patients receiving iptacopan vs none of 35 receiving eculizumab or ravulizumab achieved hemoglobin ≥12 g/dL (P <.0001). Fifty-one of 60 iptacopan-treated patients had ≥2 g/dL hemoglobin increase vs baseline compared with none of those receiving standard of care (P < .0001).15

APPLY-PNH: Impact of Iptacopan on Fatigue
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
As we know, patients with ongoing anemia often continue to suffer from significant fatigue that can be debilitating at times. The treatment with iptacopan was associated with an improvement in FACIT-Fatigue score that often approached that of the general population. Overall, in this trial, improvements in hemoglobin and fatigue ran essentially in parallel with each other.15

APPULSE-PNH: Iptacopan in Patients With PNH Who Switched From Current SoC
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
The results of the APPULSE-PNH trial were reported at the European Hematology Association meeting in 2025.16 In this international, single-arm, open-label phase IIIb trial, patients with PNH and hemoglobin ≥10 g/dL on stable anti-C5 therapy without RBC transfusions for at least 6 months were treated with oral iptacopan 200 mg twice daily for 24 weeks. The study was designed to explore whether switching to iptacopan can increase hemoglobin in these patients, who, by some standards, would be considered to have their PNH under reasonable control.
The primary endpoint was the change in hemoglobin levels from baseline. Key secondary endpoints included the proportion of hematological responders, the change in ARC levels and LDH levels from baseline, and safety (NCT05630001).16

APPULSE-PNH: Study Outcomes
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
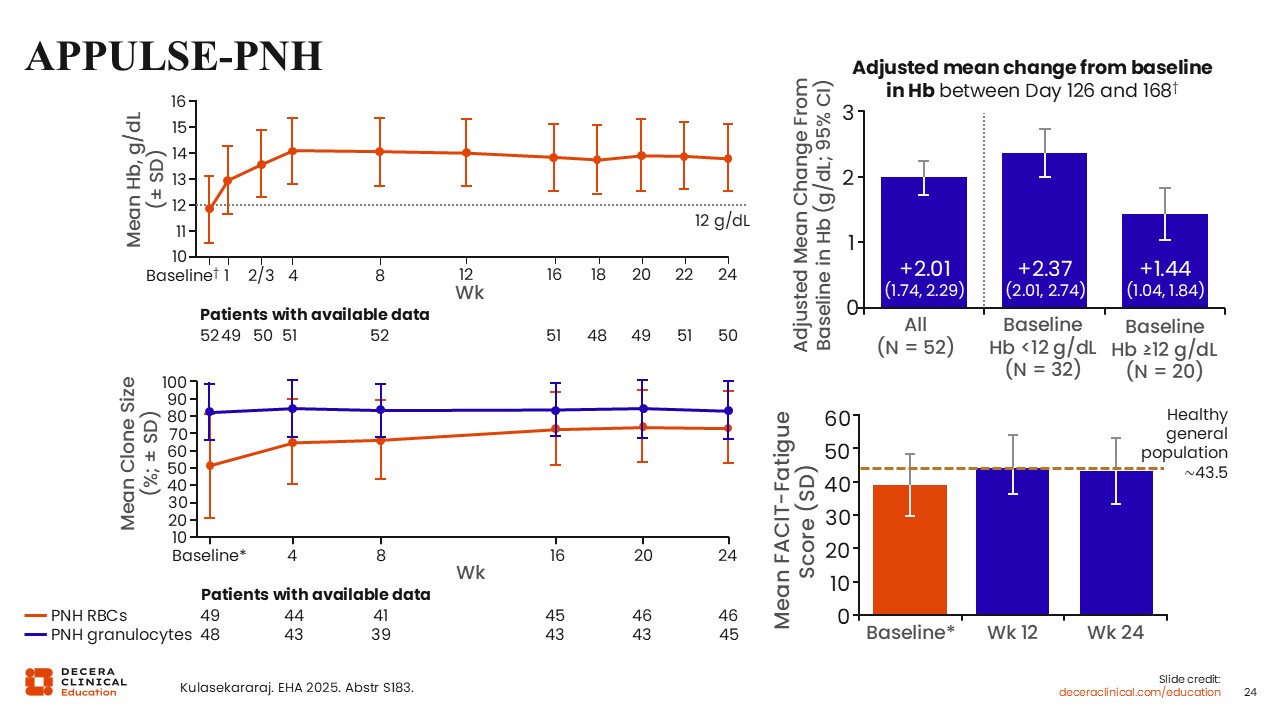
Despite starting the study with hemoglobin above 10 g/dL, many of these patients experienced an improvement in hemoglobin following 24 weeks of iptacoplan.16 The adjusted mean change in hemoglobin overall was 2.01 g/dL, and iptacopan met prespecified criteria for noninferiority and superiority to previous anti-C5 therapy (P <.0001). In patients with baseline levels <12 g/dL, mean increase in hemoglobin was 2.37 g/dL and 1.44 g/dL in patients with higher baseline levels.
Mean ARC was reduced, and there was no breakthrough hemolysis or need for transfusions. Further evidence for control of hemolysis can be seen in the figure on the left lower part of the slide, where we can see that the clone size, based on red cells, very closely approached that of monocytes and granulocytes, showing that these red cells are now protected from both intravascular and extravascular hemolysis and are essentially maintaining their normal lifespan. On the right lower part of the slide, the FACIT-Fatigue score essentially became identical to that of the general population, indicating a significant improvement in quality of life for these patients.16
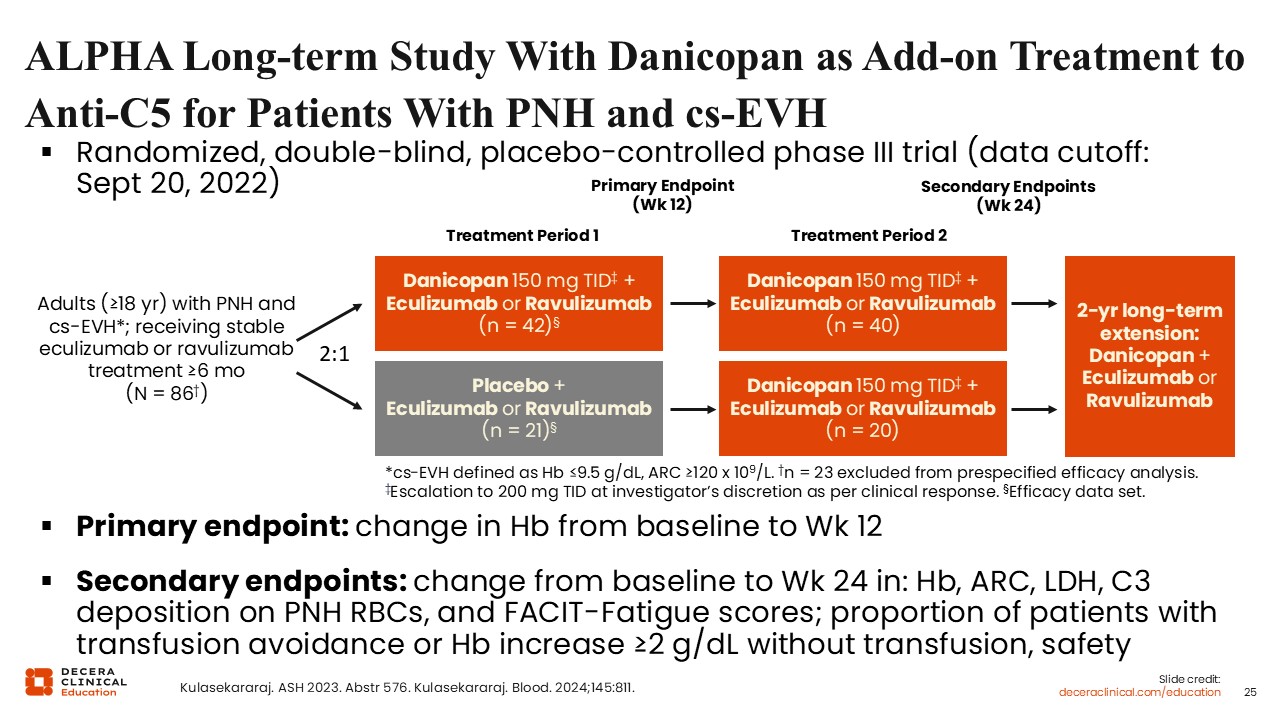
ALPHA Long-term Study With Danicopan as Add-on Treatment to Anti-C5 for Patients With PNH and cs-EVH
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
Danicopan is an oral Factor D inhibitor that is approved in the United States as an add-on therapy to ravulizumab or eculizumab for the treatment of extravascular hemolysis in adults with PNH.17 Approval was based on findings from the ALPHA study, a randomized, double-blind, placebo-controlled phase III trial conducted in adults with PNH and clinically significant extravascular hemolysis on stable eculizumab or ravulizumab for at least 6 months, that assessed the impact of simultaneous proximal and terminal complement inhibition in these patients.18,19
Participants were randomized to add danicopan 150 mg 3 times daily or placebo to baseline therapy for an initial 12-week period, followed by a further 12 weeks of danicopan plus baseline therapy in all patients, and a 2-year extension phase. The primary endpoint was the change in hemoglobin from baseline to Week 12. Secondary endpoints included the change from baseline to Week 24 in hemoglobin, ARC, LDH, C3 deposition on PNH RBCs, and FACIT-Fatigue scores; the proportion of patients with transfusion avoidance or ≥2 g/dL hemoglobin increase without transfusion, and safety.18,19
ALPHA: Hemoglobin Levels Over Time
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
Patients randomized to immediate addition of danicopan had a rapid increase in hemoglobin. At Week 12, the least-squares mean increase in this arm was 2.94 g/dL compared with 0.50 g/dL in the placebo arm. Patients who were randomized to placebo and added danicopan at Week 12 similarly experienced a rapid improvement in hemoglobin, with a least-squares mean increase of 2.26 g/dL at Week 24. Both groups maintained the improvement in hemoglobin levels through Week 48.19

ALPHA: FACIT-Fatigue Scores After Week 12 With Danicopan as Add-on Treatment
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
FACIT-Fatigue scores improved with addition of danicopan as hemoglobin levels increased.18 At Week 72, FACIT-Fatigue scores approached the general population normal level in the immediate danicopan group. Patients in this arm remained transfusion independent, whereas the need for transfusion was reduced in the placebo arm from Week 12 to Week 24.18,19

Crovalimab: Properties
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
In contrast to eculizumab and ravulizumab, which are given intravenously, crovalimab was developed as a monoclonal antibody against C5 that can be given subcutaneously.20 Crovalimab binds to wild-type C5 and a C5 with a single missense mutation or single nucleotide polymorphism that is associated with a lack of response to eculizumab. Other properties of crovalimab includes its ability to undergo recycling, which extends its half-life. As a result, the antibody can be given subcutaneously every 4 weeks, after a 28-day loading-dose period.
As shown, switching from eculizumab or ravulizumab to crovalimab can result in the generation of drug–target–drug complexes (DTDCs). During simultaneous antibody exposure, crovalimab and eculizumab bind to different epitopes on C5, allowing formation of circulating DTDCs. These may result in a serum sickness-like state that is typically associated with the first dose of therapy and should resolve with time.

COMPOSER: 3-Part Adaptive Phase I/II Trial of Crovalimab in Healthy Volunteers and Patients With PNH
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
The COMPOSER trial was a 3-part adaptive phase I/II trial of crovalimab in healthy volunteers and patients with PNH.20 The study was designed to assess the safety, tolerability, and pharmacodynamics of crovalimab in both populations and explore efficacy and immunogenicity in patients with PNH who were either naive to therapy or pretreated with eculizumab.
This study found that crovalimab was tolerated with acceptable safety. DTDC was observed in a small number of patients but was transient and did not affect efficacy.

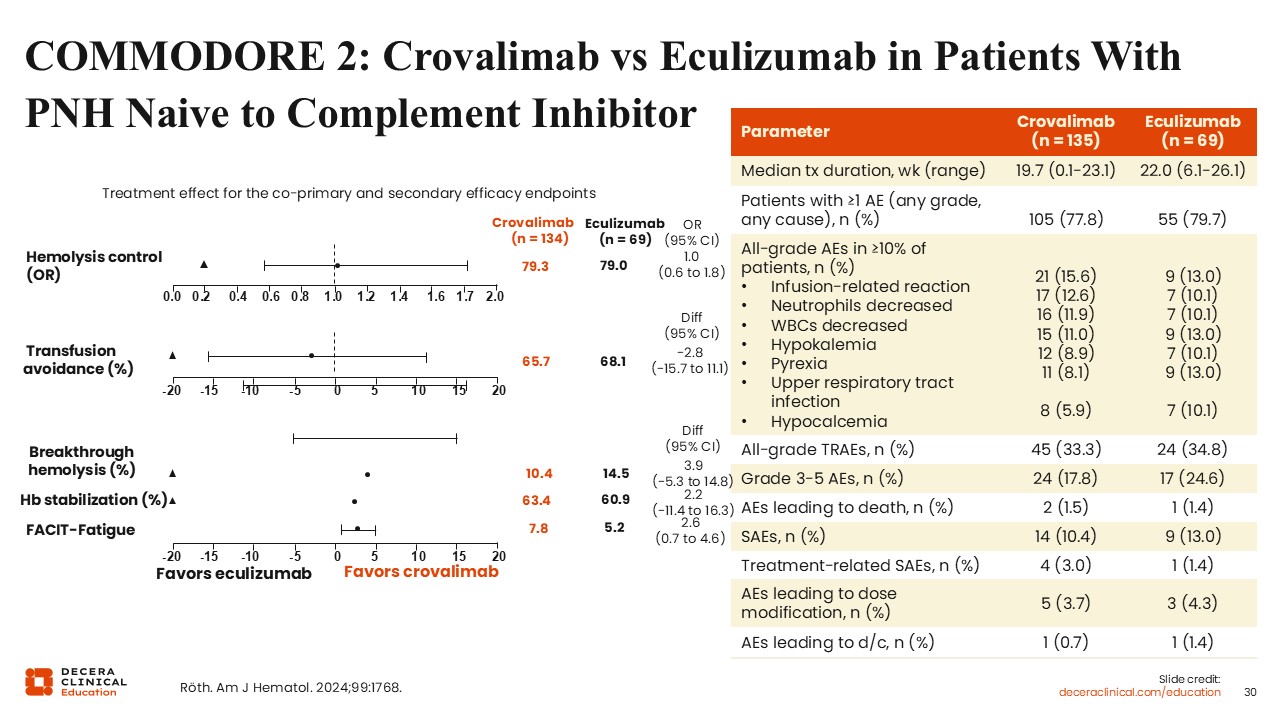
COMMODORE 2: Crovalimab vs Eculizumab in Patients With PNH Naive to Complement Inhibitor
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
COMMODORE 2 was a multicenter, randomized, open-label phase III trial evaluating the noninferiority of crovalimab vs eculizumab in complement inhibitor–naive patients with PNH.21 The coprimary endpoints were hemolysis and transfusion avoidance. Secondary endpoints included breakthrough hemolysis, stabilized hemoglobin, and FACIT-Fatigue score.
The primary treatment period was 24 weeks, and crovalimab met prespecified criteria for noninferiority to eculizumab regarding hemolysis and transfusion avoidance at this timepoint. The overall safety profiles of the 2 agents were similar.
These data show that eculizumab or crovalimab are more or less equivalent, the difference between them being the route of administration and the frequency of dosing. In principle, crovalimab could be given in the patient’s home and perhaps even self-administered, although that is not currently an approved strategy in the United States.22
Complement Inhibition in PNH
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
Despite PNH being a rare disease, we are fortunate to have multiple complement inhibitors available, that are approved either alone or in combination, and these have transformed the clinical landscape for our patients.23 Proximal inhibitors, as we have discussed, have improved disease control further, although breakthrough hemolysis remains a challenge.
Several novel agents are under investigation. These include the anti-C5 combination of pozelimab and cemdisiran; zaltenibart, an MASP-3 inhibitor that targets the lectin pathway; and KP104, a bifunctional fusion protein designed to block intravascular and extravascular hemolysis by targeting both C5 and C3.23

Phase II Trial of Pozelimab and Cemdisiran in Patients With PNH
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
The rationale for combining pozelimab and cemdisiran is that it should provide C5 suppression at reduced doses and with a longer dosing interval, potentially allowing for self-administration.
The combination of pozelimab and cemdisiran was investigated in an open-label, randomized phase II trial.24 This study was conducted in adults with PNH who had received pozelimab monotherapy in the open-label extension trial (NCT04162470) and were willing to switch to the combination of pozelimab and cemdisiran. Patients were randomized to one of 2 pozelimab and cemdisiran subcutaneous dosing regimens for a 28-week treatment period: Pozelimab was given either every 2 weeks or every 4 weeks, with cemdisiran every 4 weeks. The primary objective was to assess safety and tolerability through the randomized treatment phase.

Phase II Trial of Pozelimab and Cemdisiran: Outcomes
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
The combination was generally well tolerated, and most treatment-emergent adverse events were mild to moderate.24 The combination was effective at blocking hemolysis and improved hemoglobin, FACIT-Fatigue score, and physical functioning. The frequency of pozelimab dosing did not affect disease control, and the every-4-week dosing regimen is being further studied in phase III trials. For example, several studies with this therapeutic combination are currently recruiting (NCT07154745, NCT05133531, and NCT05744921).

Phase II Trial of Tesidolumab in Patients With Variant and Nonvariant C5: Key Clinical Endpoints
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
Tesidolumab is another monoclonal antibody that targets C5.25 Polymorphisms in C5 render eculizumab and ravulizumab ineffective in a subset of PNH patients who harbor this variant. Tesidolumab attaches to a different C5 epitope and, in principle, should be effective in such a patient population. This approach has been tested in an open-label, single-arm phase II trial of tesidolumab in patients with variant and nonvariant C5. Participants received tesidolumab for an initial 4-week period, followed by optional continued treatment through Week 48. The primary endpoint was LDH reduction at Day 29, with secondary endpoints focused on safety, tolerability, and pharmacokinetics. Treatment with tesidolumab resulted in near normalization of LDH in patients with either variant and nonvariant C5 and was well tolerated overall. Hemoglobin levels increased, but not optimally, suggesting additional therapy may be needed.

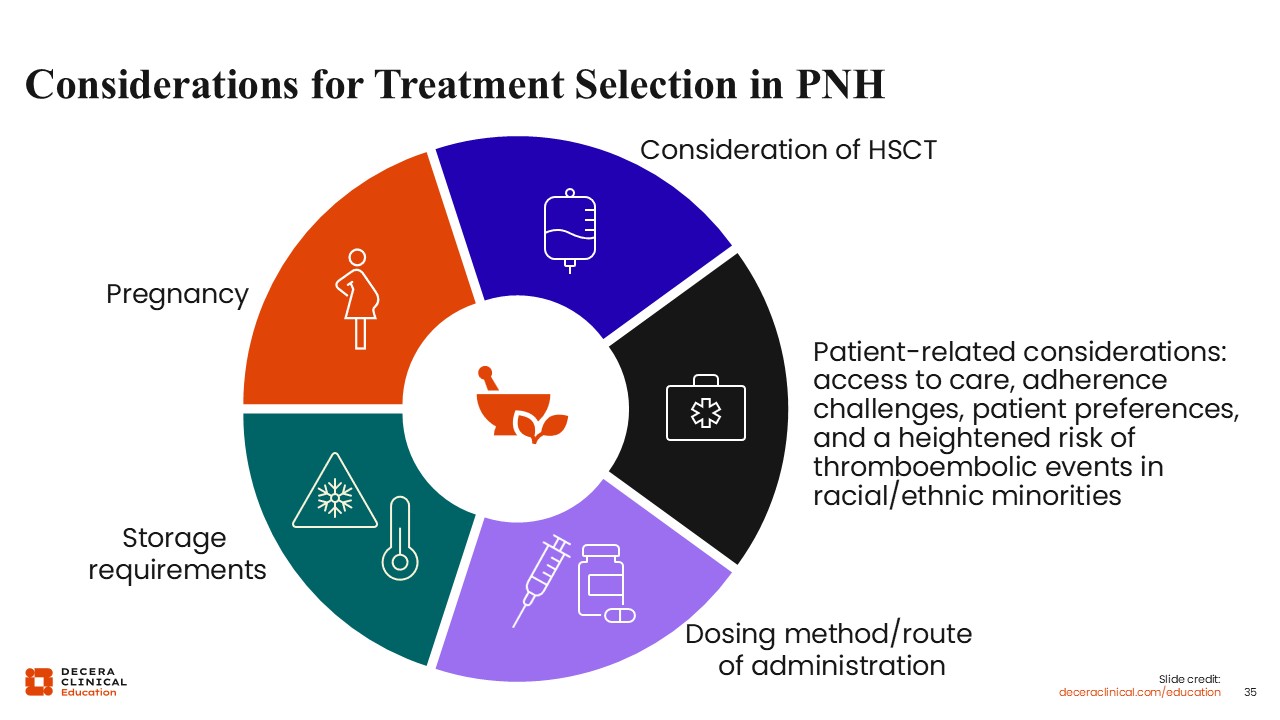
Considerations for Treatment Selection in PNH
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
Choosing therapy for patients with PNH is based on consideration of various medication-related and patient-related factors. Decision-making should involve the patient because there is no one best approach. In my practice, this involves talking with the patient about the method of administration (intravenous or subcutaneous vs an oral agent), the dosing frequency, and any storage requirements. For example, pegcetacoplan needs to be stored in a refrigerator. Where would patients access their therapy? Are patients willing to self-administer the product at home? Would they like a nurse to visit them at home and infuse them with the product? Or would they prefer an oral agent that they can take in the comfort of their home?
Potential complications should also be considered, including the risk of thromboembolic events and the likelihood of compliance with therapy. For younger women with PNH, how to choose and manage therapy alongside potential pregnancy should also be discussed.
Transplantation remains the only curative therapy for PNH but is not generally recommended in classic hemolytic PNH given the efficacy of current therapeutic approaches.
Allogeneic Transplant for PNH
David Dingli, MD, PhD, FRCP, FRCPEd, FACP, FRCPath:
I typically consider an allogeneic transplant for patients with PNH who have an associated bone marrow failure syndrome, typically, aplastic anemia. It is unusual for patients to fail complement-directed therapy, but that would also signal consideration of an allogeneic transplant. The risk of thrombosis is substantial in these patients. If there is a history of recurrent thrombosis despite adequate complement-directed therapy, some healthcare professionals will consider an allogeneic stem cell transplant as a therapeutic option.

Addressing the Burden of Disease in Patients With PNH
Carlos M. De Castro, MD:
For the remainder of this module, I focus on the burden of disease in patients with PNH and how this can be addressed. Physical burdens on the patient include their health status, their vaccine status, and concerns about breakthrough hemolysis.26 There can be problems with treatment, including diagnostic delays, problems with insurance coverage, adverse events, and how to weigh decisions about which treatment is best for each patient. These can result in emotional stress, and for many patients, PNH has financial consequences through the impact on work and the cost of medication and care.

Challenges of Delayed Diagnosis of PNH
Carlos M. De Castro, MD:
Diagnosis of PNH is often delayed because it is a rare disease, and many healthcare professionals will not have encountered PNH before. Patients may present with multiple nonspecific, unexplained symptoms.27 Fewer than 40% of patients with PNH receive their diagnosis within 12 months of symptom onset.28 The average delay is approximately 2 years from initial symptoms and can be as long as 5 years for some patients. Most patients will see more than 1 healthcare professional—typically urologists, cardiologists, gastroenterologists, pulmonary doctors, and neurologists—before they are diagnosed.

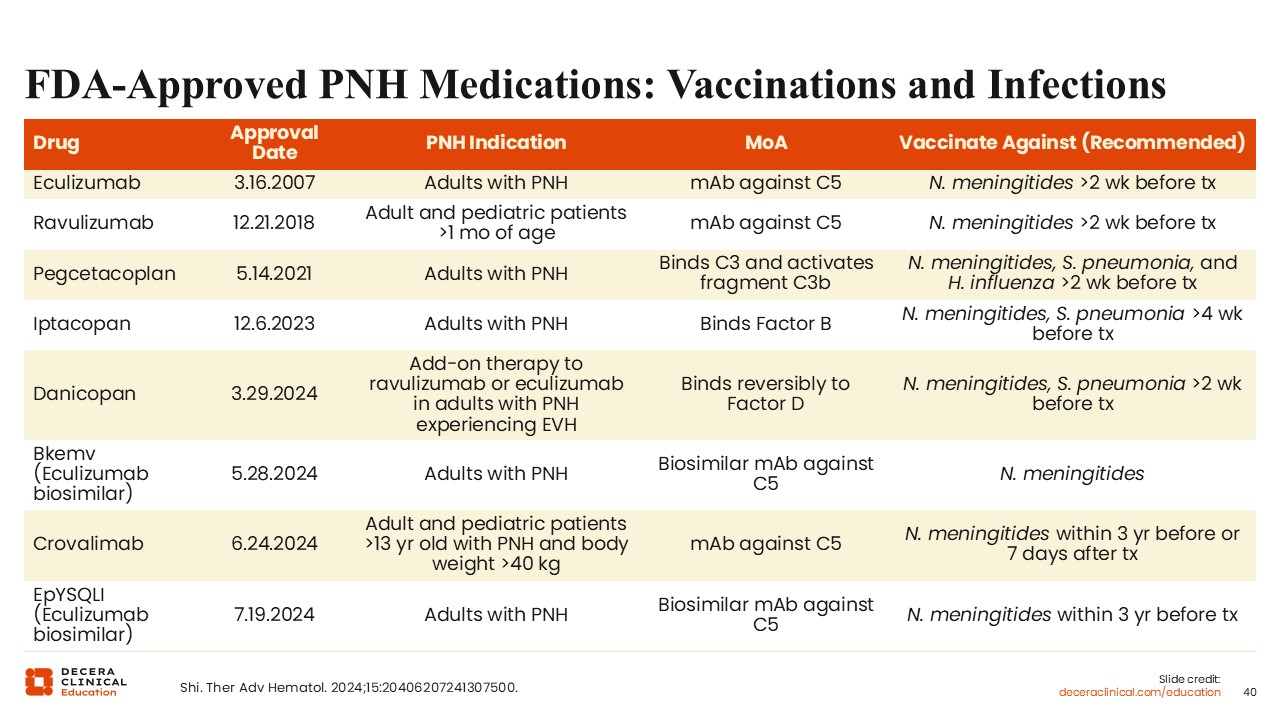
FDA-Approved PNH Medications: Vaccinations and Infections
Carlos M. De Castro, MD:
All complement inhibitors used to treat PNH are associated with an increased risk of infections, including meningococcal meningitis, other bacterial infections, and viral infections.1 Complement inhibitors carry a boxed warning about the risk of meningococcal infections and must be prescribed under a Risk Evaluation and Mitigation Strategy program. Vaccination against meningococcus must be completed or updated prior to starting treatment, according to the schedule recommended for each therapy, usually at least 2 weeks before treatment is initiated. If starting complement inhibitor therapy is urgent, prophylactic antibiotics should be administered and vaccination given as soon as possible. For patients beginning proximal inhibitors (pegcetacoplan, iptacopan, danicopan), vaccination against encapsulated organisms such as Streptococcus pneumoniae and Haemophilus influenzae should be administered using Advisory Committee on Immunization Practices guidelines.
It is important to remember that vaccines are not 100% effective. Patients need to be educated about signs and symptoms of meningitis and other infections and to seek immediate medical attention if these occur.
It is important to be aware that vaccines can activate complement and lead to a hemolytic flare in patients who are not receiving a complement inhibitor. One strategy to prevent flares from happening is to start prophylactic antibiotics, then give the complement inhibitor, and then administer the vaccine(s) shortly afterwards.
Managing Treatment-Related Adverse Events Associated with Complement Inhibitors in PNH
Carlos M. De Castro, MD:
Treatment-related adverse events seen in patients receiving complement inhibitors include fatigue, anemia, iron deficiency, injection site reactions, diarrhea, and hyperlipidemia, and certain drugs, including proximal inhibitors, present risk of thrombosis.1 As discussed, switching from one C5 inhibitor to another can result in transient immune complexes, which usually cause a rash.

Persistent Anemia in PNH
Carlos M. De Castro, MD:
Patients with persistent anemia on PNH treatment require a workup to identify the cause. It is common for patients receiving a C5 inhibitor to be anemic, and some may require transfusions. Anemia may be caused by iron or other nutritional deficiency. There may be associated bone marrow failure, which can be determined by a reticulocyte count. Patients with PNH may have comorbid inflammation or chronic kidney disease that may or may not be related to PNH, which can also cause anemia.
Patients receiving complement inhibitors may experience persistent intravascular hemolysis. Patients with breakthrough hemolysis on a C5 inhibitor should have CH50 trough level, LDH, and bilirubin measured. Extravascular hemolysis is usually marked by an elevated reticulocyte count and mildly elevated bilirubin, and the Coombs test may be positive for C3. Usually, LDH is mildly elevated, and flow cytometry may detect C3 fragments on RBCs.

Disease Burden: Breakthrough Hemolysis in PNH
Carlos M. De Castro, MD:
Breakthrough hemolysis in patients with PNH is commonly described as the sudden return of symptoms related to intravascular hemolysis, such as dark urine or hemoglobinuria, with a significant rise in LDH levels and a sharp drop in hemoglobin, with or without any major adverse vascular events.
There are multiple causes of breakthrough hemolysis in patients with PNH.29 Untreated patients commonly have intravascular hemolysis that is mediated through the membrane attack complex. In patients receiving a C5 inhibitor, RBCs can become coated with C3 fragments (opsonized), causing them to be taken up in the liver and the spleen, leading to extravascular hemolysis. Pharmacokinetic breakthrough hemolysis occurs when the patient’s drug levels fall below that needed for adequate C5 inhibition, usually seen at the end of the dosing period prior to whenever the next dose is due. Pharmacodynamic breakthrough hemolysis occurs where there is a complement-activating event, such as an infection, that overwhelms the complement inhibitor. Management of this type of breakthrough hemolysis usually involves treating the complement-activating event, for example, giving antibiotics for infections, and possibly giving a different complement inhibitor.

Managing Breakthrough Hemolysis in PNH
Carlos M. De Castro, MD:
Breakthrough hemolysis requires prompt identification and management. Healthcare professionals and their patients should be aware of the signs and symptoms of breakthrough hemolysis, including dark urine or a return of other PNH symptoms. Prevention strategies, such as giving antibiotics for home or even prophylactic use, may be considered, but this is not universally accepted, and recommended management strategies for breakthrough hemolysis are lacking.30 In severe cases, giving another dose of the complement inhibitor or switching to a different inhibitor are common approaches.

Pregnancy and PNH
Carlos M. De Castro, MD:
Pregnancy in patients with PNH is an important clinical challenge.31 Prior to complement inhibitor therapy, patients with PNH who became pregnant ran a high risk of miscarriages and fetal demise, in addition to maternal demise, usually from a thrombotic event. We recommend, therefore, that pregnancy in a patient with PNH should be planned carefully in conjunction with their healthcare professionals. Most of the prior clinical experience using complement inhibitors in pregnancy is with eculizumab, which has been shown to improve maternal and fetal outcomes.32 Data on other therapies are limited. Patients with PNH who become pregnant or are planning pregnancy are commonly given eculizumab. Dose adjustments may be needed to keep the drug at a therapeutic level because of the increased volume during pregnancy. Anticoagulation is recommended from approximately the second trimester until postpartum to prevent clots. Finally, obstetricians with expertise in high-risk pregnancy should manage obstetric care alongside a PNH expert.

Communication: Patient Discussions and Strategies to Address Disease Burden of PNH
Carlos M. De Castro, MD:
It is important to discuss these strategies for addressing the disease burden of PNH with patients themselves. This might include how we manage fatigue and other symptoms, risks and management of treatment-related adverse effects, how to support mental health through stress mitigation, and access to counselors. Finally, there is the issue of financial problems because of the cost of the drugs and whether insurance will cover them.
Education for Patients and Their Caregivers: Patient Resources
Carlos M. De Castro, MD:
Educational resources available for patients with PNH and their caregivers are shown in the slide. These include the Aplastic Anemia and MDS International Foundation, a group that also deals with PNH; the National Organization for Rare Disorders; and the PNH Global Alliance. The website, myPNHteam, provides information about PNH and how to manage different problems. In addition, groups like the Aplastic Anemia and MDS International Foundation hold regional meetings for patients and have a wealth of information on PNH on their website.
In summary, we hope this has been an informative coverage of PNH. Starting with the pathophysiology of disease, there is usually a lengthy delay in diagnosing patients. Flow cytometry can be very helpful for an accurate diagnosis, and now there are many therapeutic options for treating this disease. No longer do patients need to suffer with anemia and the overwhelming disease burden. There are also new drug approvals that improve first-generation agents and ongoing clinical trials for additional potential therapies. It is important to find the right treatment and have conversations with healthcare professionals to enhance quality of life for patients with PNH.
