CME
Diagnostic Precision in Essential Thrombocythemia and Polycythemia Vera
Physicians: Maximum of 0.25 AMA PRA Category 1 Credit™
Released: February 02, 2026
Expiration: August 01, 2026

Activity
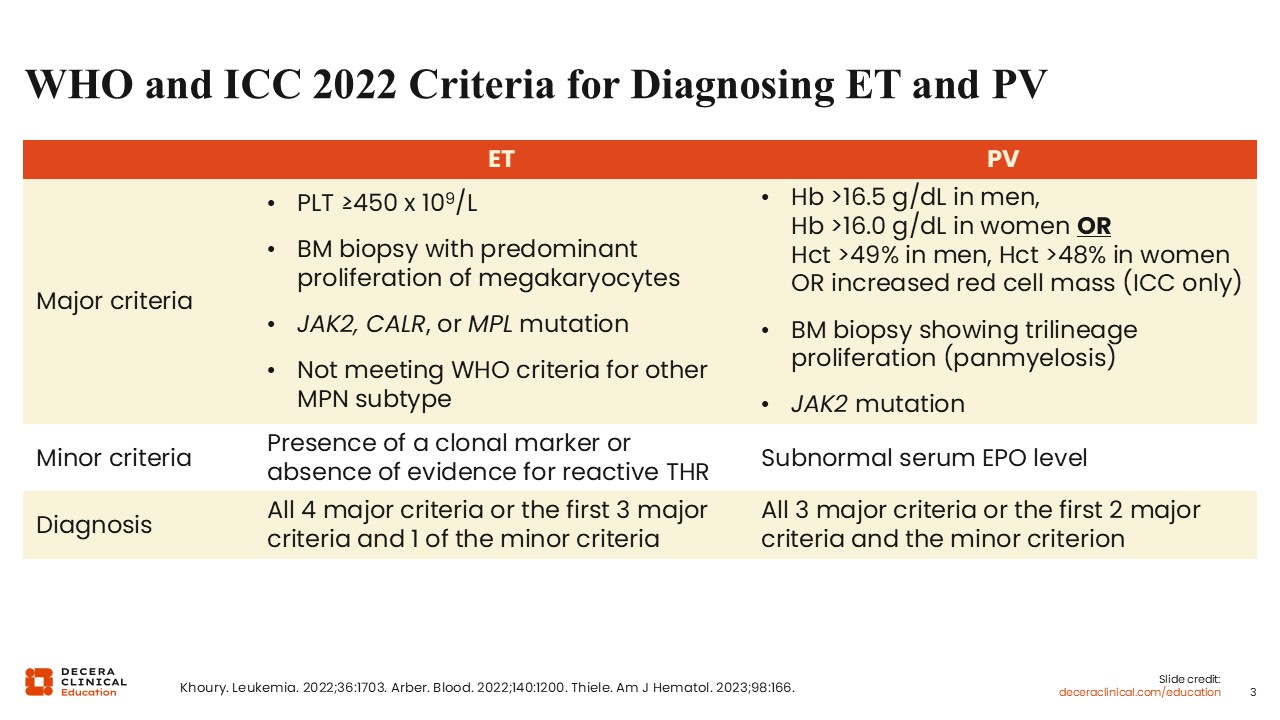
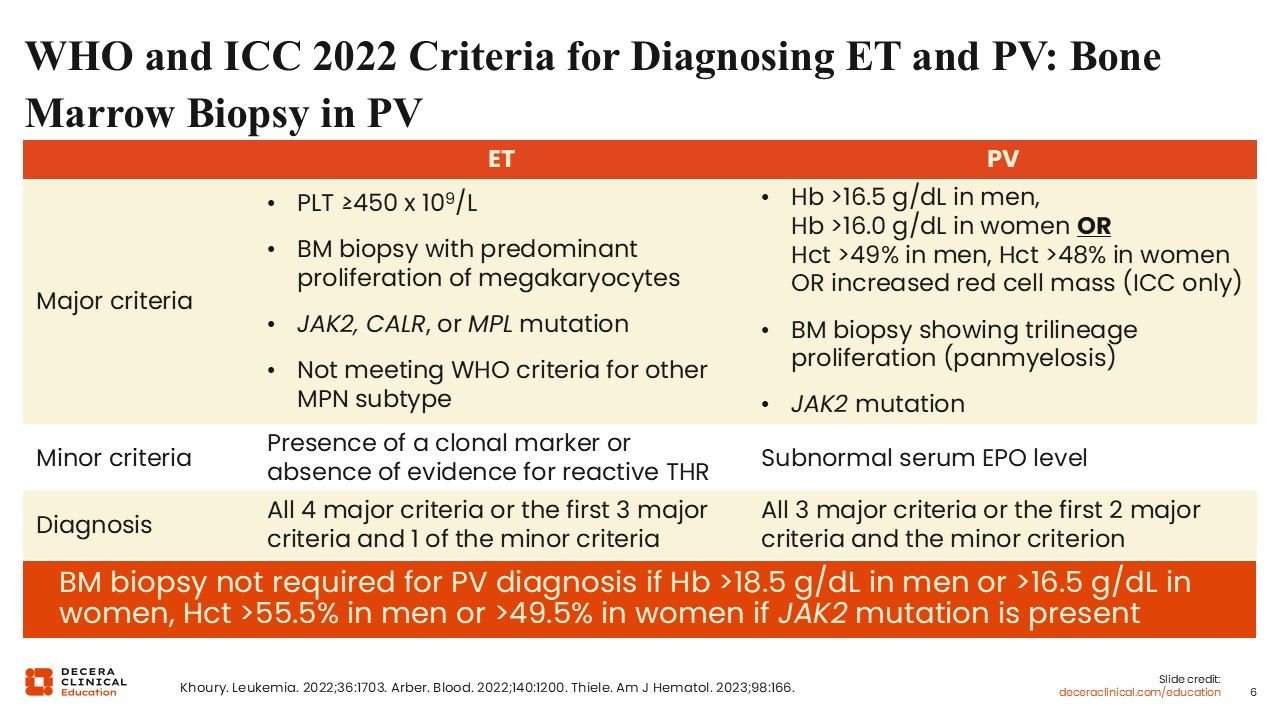
WHO and ICC 2022 Criteria for Diagnosing ET and PV
This table shows the WHO and ICC criteria for the diagnosis of ET and PV.1-3 With regards to ET, major criteria include an increased platelet count, characteristic bone marrow biopsy findings, and a major driver mutation in JAK2, CALR, or MPL. Patients cannot meet WHO criteria for any other MPN subtype. Approximately 10% of patients with ET will be triple negative, or lack a pathogenic driver mutation in JAK2, CALR, or MPL.2-4 For this reason, the minor criteria also includes a specification that there is either a nondriver pathogenic mutation, or no other clear cause of thrombocytosis. Common causes of reactive thrombocytosis include inflammation, iron deficiency, and splenectomy.3
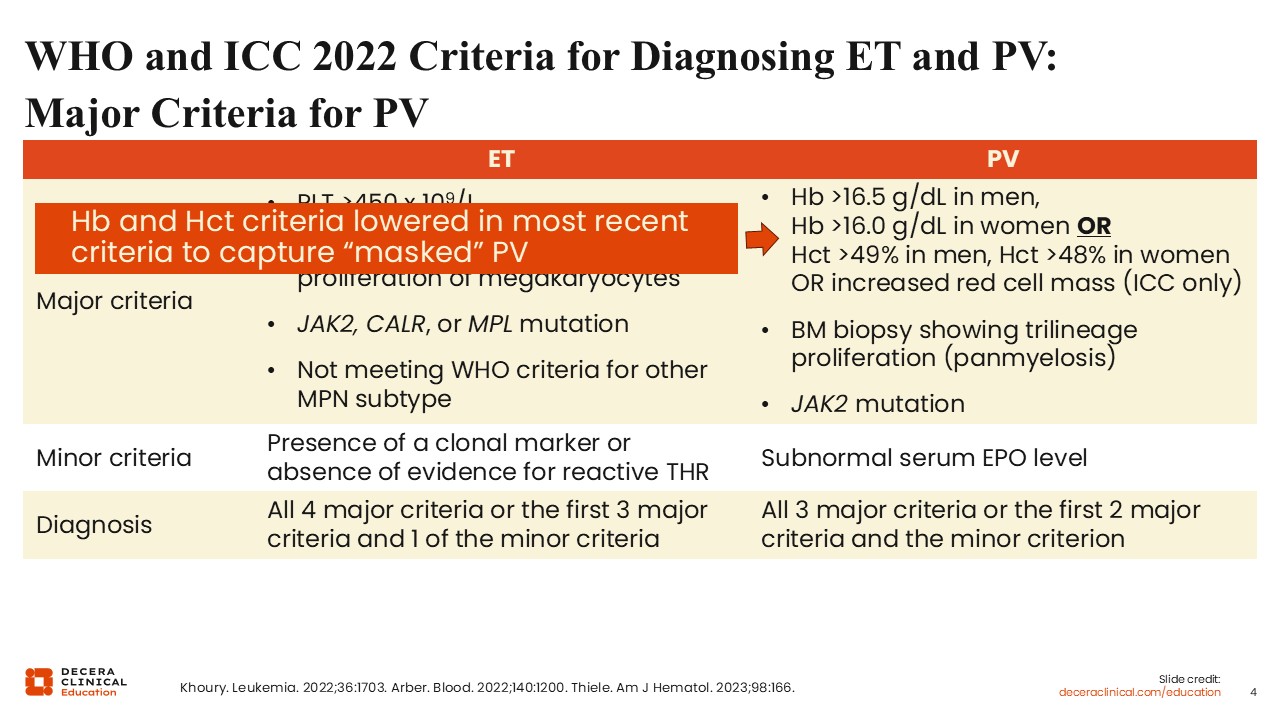
WHO and ICC 2022 Criteria for Diagnosing ET and PV: Major Criteria for PV
With regards to PV, major criteria include evidence for increased red blood cell parameters, including a hemoglobin >16.5 in men and >16 in women, or hematocrit >49% in men and >48% in women.1-3 Note that the current WHO criteria lowered the hemoglobin and hematocrit threshold in formal diagnostic criteria, in recognition that some patients with PV may not have significantly elevated red blood cell parameters.1,5 This “masked” PV is especially common in patients who present with Budd-Chiari syndrome, which is often accompanied by portal hypertension and splenomegaly.6,7 The resulting expanded plasma volume can dilute hemoglobin and hematocrit values, resulting in falsely low or “masked” phenotype. Other major criteria include characteristic bone marrow biopsy findings, more specifically panmyelosis as PV is characterized by myeloid, not just erythroid, expansion, and a JAK2 mutation, which occurs in nearly all patients with PV.
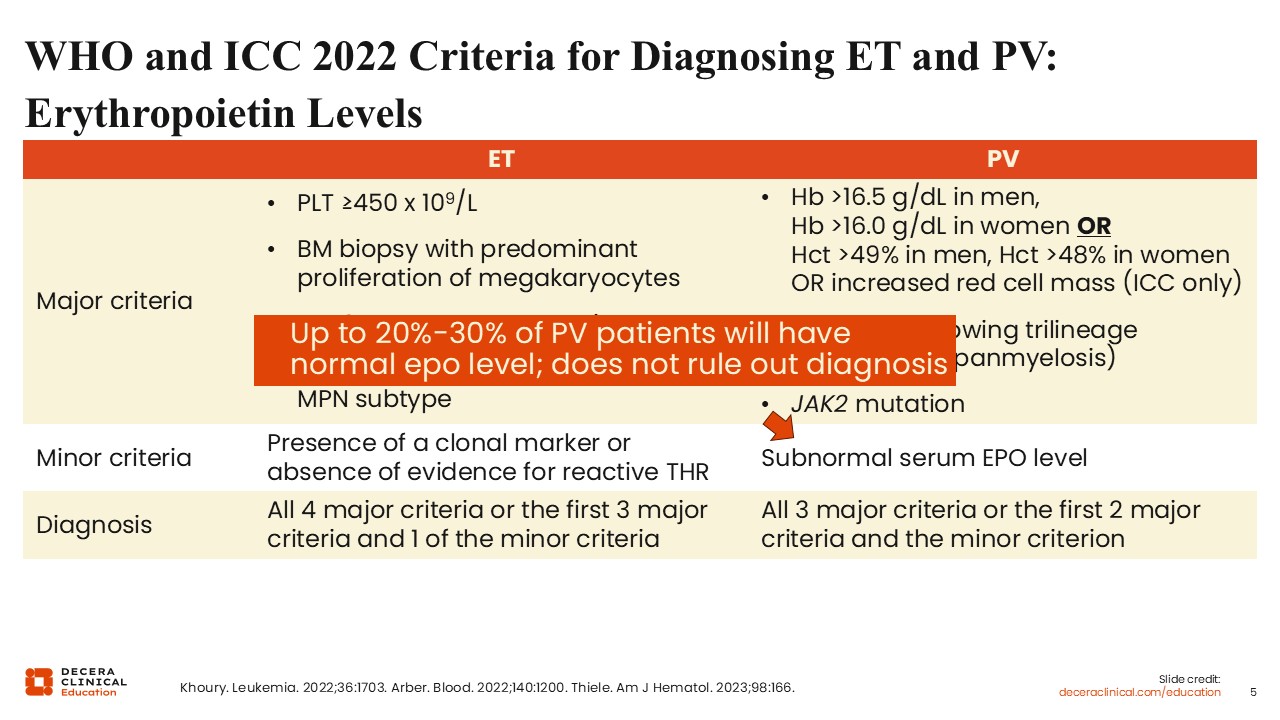
WHO and ICC 2022 Criteria for Diagnosing ET and PV: Erythropoietin Levels
A subnormal erythropoietin level is a minor criterion for the diagnosis of PV, although it is important to note that up to 20% to 30% of patients with PV will have a normal erythropoietin level, and many can even have increased erythropoietin levels.1-3,8 A normal erythropoietin level does not rule out the diagnosis of PV, but a low erythropoietin level in a patient with erythrocytosis awaiting genetic marker testing is strongly suggestive of PV.
WHO and ICC 2022 Criteria for Diagnosing ET and PV: Bone Marrow Biopsy in PV
Of note, the WHO and ICC criteria do not require a bone marrow biopsy for PV diagnosis if a patient has sustained erythrocytosis, a JAK2 mutation, and if they also have a subnormal erythropoietin level.2,8-10 In practice, a diagnostic bone marrow biopsy can still be helpful for establishing a baseline and providing some prognostic information, particularly in younger patients who are at risk of having progression occur in their lifetime.
WHO and ICC 2022 Criteria for Diagnosing ET and PV: Bone Marrow Biopsy in ET
By contrast, by both WHO and ICC criteria, a bone marrow biopsy is required to make a diagnosis of ET, mostly as it is needed to differentiate ET from other MPNs that can also present with isolated thrombocytosis, such as prefibrotic myelofibrosis (MF) or less commonly chronic myeloid leukemia.1-3 A bone marrow biopsy is also particularly helpful in differentiating reactive thrombocytosis from triple-negative thrombocytosis. There is some practice variation with regards to whether healthcare professionals do obtain a bone marrow biopsy to formally diagnose ET, given that practically, the bone marrow biopsy infrequently changes management. For instance, in a patient with a JAK2 mutation and thrombocytosis who is found to have pathologic findings that favor prefibrotic MF in the bone marrow, they often clinically behave more like ET and so are treated as ET. However, I tend to favor a diagnostic bone marrow biopsy as a baseline, even though management often does not change, the biopsy can add prognostic information. In frailer or older patients, or those who are strongly against a bone marrow biopsy, it may be more practical to omit. However, if there are concerns for progression, including to overt MF, patients should be counseled that a bone marrow biopsy is more strongly indicated.

Overlapping Features With Essential Thrombocythemia, Prefibrotic Myelofibrosis, and Polycythemia Vera
It can be challenging to make a diagnosis of ET, PV, and prefibrotic MF in part because these diseases often exist on a spectrum.1-3,11 Patients with ET may progress to PV, with rising JAK2 levels over time and increased red blood cell parameters, before progressing to overt MF. When you first see a patient and get a bone marrow biopsy, you are capturing them at 1 moment in time, and so sometimes it can be difficult to distinguish the diagnoses. In practice the management of ET and MF is similar. Even if a patient carries an “ET” diagnosis with a JAK2 mutation, you would still want to keep their hematocrit less than 45% to reduce thrombotic risk. Prefibrotic MF can also be difficult to distinguish from ET.12 This is a relatively newer entity that was formally defined in the 2016 WHO criteria with specific histopathologic criteria, distinct from overt PMF and ET.13 However the pathologic criteria that differentiates the 2 can be subtle, and the distinction is almost decided by morphology, especially megakaryocyte appearance and background cellularity. This is complicated by the fact that patients may sit at a continuum, and the bone marrow itself may be patchy. Clinically, many patients with prefibrotic MF will look similar to patients with ET as well.

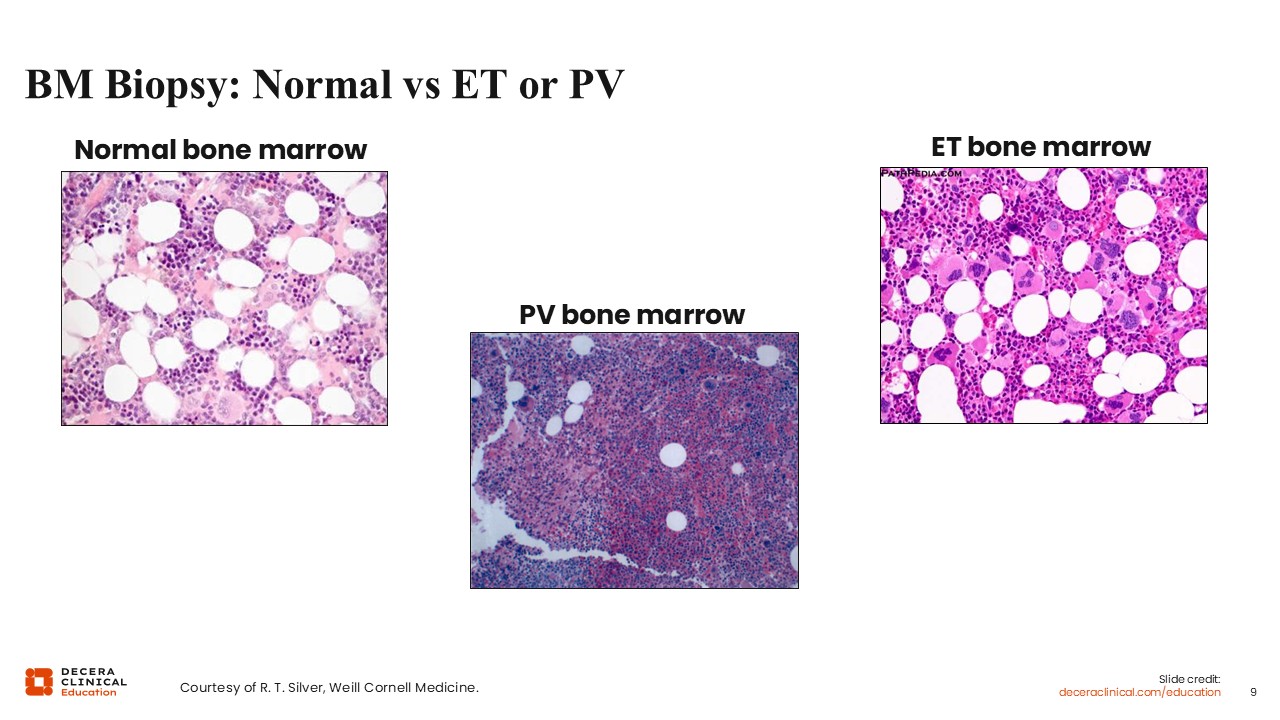
BM Biopsy: Normal vs ET or PV
This slide captures some of the key differences in bone marrow morphology between ET, PV, and normal. Patients with ET should have a normocellular marrow, and a hypercellular marrow should tip you off to a PV or even prefibrotic MF diagnosis.1-3 You can see the differences in cellularity based on the fat present in the bone marrow biopsies, with PV having more blue and less fat compared to the normal and ET bone marrow. In PV, there is trilineage hyperplasia, meaning that all 3 lineages are expanded, including erythroid, granulocyte, and megakaryocytes, which is reflected in the dense cellular background. In contrast, ET is primarily a megakaryocytic disorder, with increased megakaryocytes. Many megakaryocytes are large to giant, clustered, and hyperlobated, with “staghorn like nuclei” which you can see in the large cells in the center of the ET picture. In both ET and PV, there should be minimal reticulin, as a grade 2 or grade 3 fibrosis grading is a requirement of overt MF. But again, diagnosis can be challenging, as patients may be actively evolving, and the inherent patchiness of the bone marrow may also lead to some misdiagnosis.
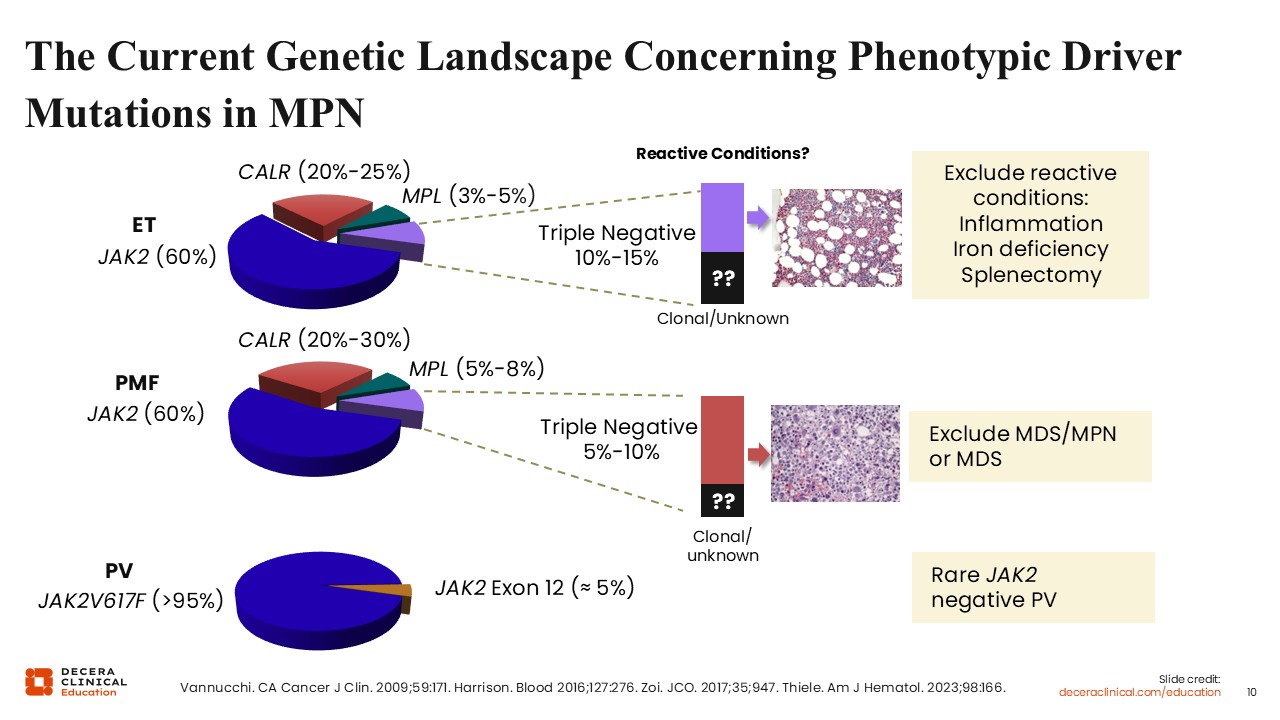
The Current Genetic Landscape Concerning Phenotypic Driver Mutations in MPN
Understanding the genetic landscape of MPNs is important for diagnosis. In ET, approximately 90% of patients will have a pathogenic driver mutation in JAK2, CALR, or MPL.4 JAK2 mutations comprise the majority; they are found in approximately 60% of patients, most commonly JAK2 V617F. CALR mutations are the most common non-JAK2 mutation, present in 80% of JAK2-negative patients and comprising 20% of all patients with MPN.14,15 MPL mutations are found in a minority (3%-5%) of patients.4 Patients without any of these common mutations are known as triple negative.16 The triple-negative patients are a heterogenous group. They generally should have a clonal marker and demonstrate other mutations outside of JAK2, CALR, and MPL, although this is not always the case. Historically patients with triple-negative ET have demonstrated good prognosis, but this is likely because retrospective studies have mistakenly characterized some patients with reactive or even familial thrombocytosis as a triple-negative ET. Besides looking for reactive conditions, the pattern of thrombocytosis is important for diagnosis. Platelet counts that fluctuate widely and in particular are normal and then abnormal suggest a reactive etiology. Patients with ET or MPN typically have a more stable platelet count that rises over time. Similarly, lifelong thrombocytosis should raise the question of familial thrombocytosis, particularly if thrombocytosis is also identified in other family members.
The breakdown of driver mutations in MF is similar to ET, and in cases of patients with triple-negative MF, other myeloid disorders such as myelodysplastic /myeloproliferative neoplasms (MDS/MPN) or myelodysplastic syndrome should be ruled out.4 Patients with triple-negative MF have historically poorer prognosis because their clonal drivers are often higher risk, and they can behave more similarly to MDS/MPN overlap biology.16
The genetic landscape looks different in PV, and nearly all patients with PV have a mutation in JAK2 with 95% having a JAK2 V617F mutation.17 The remaining 5% have a JAK2 mutation in other areas in the gene, most commonly in exon 12. True JAK2-negative PV is very uncommon, although it does exist, including case reports of patients with SH2B3 mutations that lead to JAK-STAT signaling upregulation.
Not so Mutually Exclusive?
Mutations in JAK2, CALR and MPL are mutually exclusive as each mutation activates the same JAK-STAT pathway, such that having more than 1 driver in the same clone confers no biologic advantage. However, there are rare cases of patients with double- or even triple-positive MPN, most often occurring in separate clones, and with possible implications on disease trajectory.18
The presence of multiple driver mutations also brings up the question of how we handle molecular sequencing at diagnosis. In my academic practice we are fortunate to have access to a next-generation sequencing panel to concurrently test for JAK2, CALR, and MPL, and other pathogenic nondriver mutations commonly found in MPNs such as DNMT3A, TET2, or ASXL1. This allows for the detection of rare cases of multiple driver mutations. However, this may not be available in all settings, and often sequential or reflex testing is pursued, with JAK2 sent first, followed by CALR and MPL if JAK2 is negative.19 The traditional approach is to pursue reflex testing, although ultimately choice depends on test availability, cost considerations, and clinical urgency.

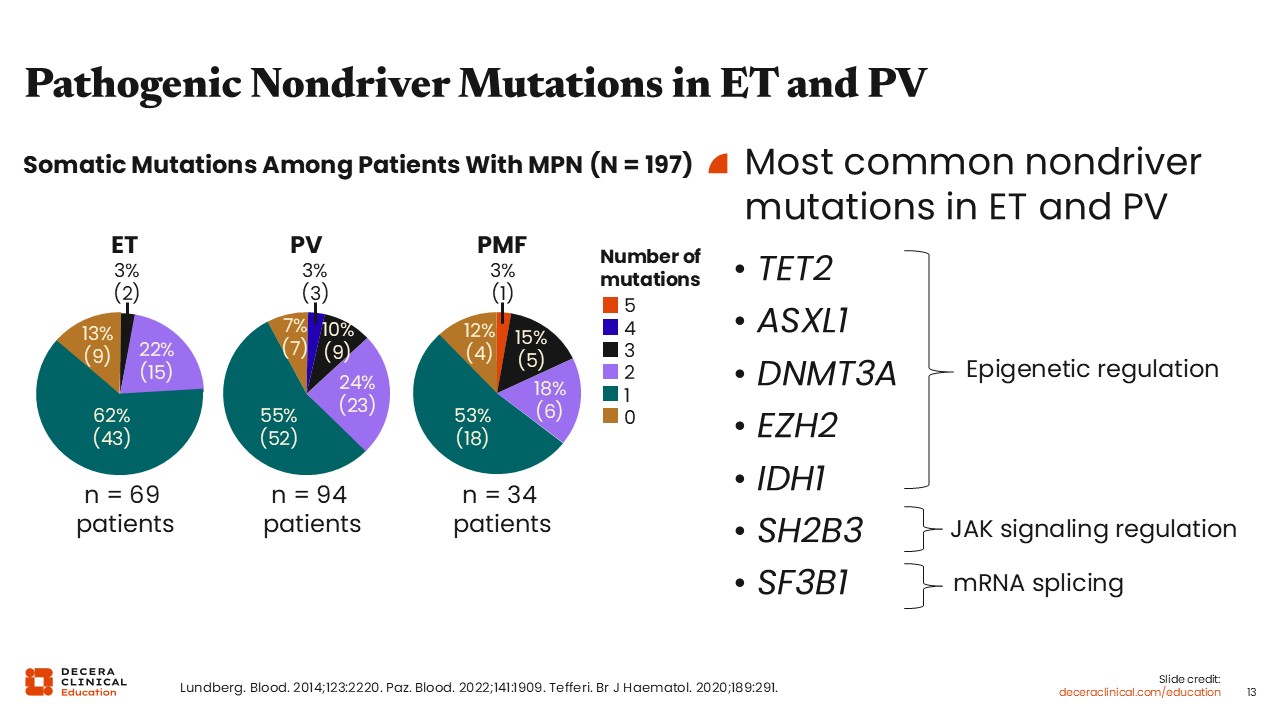
Pathogenic Nondriver Mutations in ET and PV
What is the role of genetic testing in ET and PV to identify concomitant nondriver mutations that occur? Unlike in MF where expert consensus guidelines recommend next-generation sequencing to evaluate for other nondriver mutations, the role of such testing in ET and PV is not yet clear given the lack of clinical utility of detecting these mutations at diagnosis.19 In MF, we have a plethora of scoring guidelines for prognosis that integrate molecular testing, and these results are critical for making management decisions such as transplantation.20-22 Given that there are generally fewer nondriver mutations present in ET and PV compared to MF, the evidence related to prognosis for many mutations is also less clear. However, as you can see from this 1 study, approximately 20% to 30% of patients with ET and PV will have other nondriver mutations.23 The most common mutations in ET and PV include mutations in TET2, ASXL1, and DNMT3A.
Final Thoughts on Diagnosing ET and PV
In conclusion, diagnosis of ET and PV requires patients to meet criteria for elevated blood counts and to have characteristic bone marrow biopsies.1-3 By formal criteria, a bone marrow biopsy is required for ET diagnosis but not necessary for PV diagnosis, although a diagnostic bone marrow biopsy is still helpful for prognosis and establishing a baseline of disease.2,3, Most often, however, the results of the bone marrow biopsy will not change management, so you could consider omission in patients who would not tolerate the procedure well. Diagnosis can be complicated by the fact that the MPNs lie on a spectrum, making precise classification difficult. Morphology is key, particularly for distinguishing ET from prefibrotic MF, but can have subtle findings and really requires a good pathologist to make a call. Genetic testing is also crucial for ET and PV, but unlike in MF, the value of sequencing beyond the driver mutations is less clear. How you perform molecular tests—including whether you do sequential, reflex, or concomitant testing—depends on cost, testing availability, and clinical urgency. Ongoing clinical research is needed to further refine and improve our diagnostic paradigm.

Case History
The patient is a 64-year-old man referred to you for elevated blood counts that were observed during an emergency department visit for chest pain with a subsequent diagnosis of myocardial infarction. He has a history of arterial hypertension and a deep vein thrombosis 4 years prior. He has palpable splenomegaly (2 cm). His hemoglobin is 20 g/dL, hematocrit is 61.7%, white blood cell count 12.8 x 109/L, and platelets 750 x 109/L. Additional testing reveals a JAK2 V617F positivity (VAF of 58%) and low serum erythropoietin.
Case History
A 32-year-old housewife presents with fatigue and headaches. She has splenomegaly (1 cm below costal margin). A complete blood count demonstrates platelets 650 G/L, white blood cell count 6.1 G/L, hemoglobin 13 g/dL, hematocrit 42%, normal MCV. Testing reveals normal iron and C-reactive protein, BCR-ABL1 negative, JAK2 V617F positive.